- Volume 61 , Number 2
- Page: 227–35
Molecular cloning and characterization of contiguously located repetitive and single copy DNA sequences of Mycobacterium tuberculosis: development of PCR-Based diagnostic assay
ABSTRACT
Screening of a λgt11 genomic library has been used as an approach for molecular cloning of the Mycobacterium tuberculosis repetitive DNA shown to be present on a previously described 5.6-kb Alu I genomic fragment. The recombinant clone R 18.8.2, which strongly hybridized with the radiolabeled 5.7-kb Alu fragment, carried two Eco RI inserts of 2 kb and 1.4 kb in size. Southern hybridization of each of these fragments to restriction endonucleasecleaved M. tuberculosis DNA clearly demonstrated the 2 kb to contain the repetitive DNA sequence, while the 1.4 kb is represented in a single copy. When replica plaque lifts f rom the genomic library were probed, the 5.6-kb genomic fragment and the cloned 2-kb repetitive insert hybridized to an identical number of plaques, indicating the similarity and the high copy number of the repeating unit shared by the two fragments. Restriction mapping and Southern hybridization patterns indicated that the 2-kb repetitive and the 1.4-kb single-copy DNA sequences originated f rom a contiguous piece of genomic DNA. Both fragments were found to be unique to members of the M. tuberculosis complex, except that the 2-kb insert exhibited a weak hybridization with M. kansasii DNA. Finally, a 169-bp region f rom one end of the single-copy sequence has been amplified by polymerase chain reaction (PCR) in a manner specific to members of the M. tuberculosis complex. The sensitivity of the PCR was such that as few as 9-10 bacilli could be detected.RÉSUMÉ
L'analyse d'une librairie génomique λgt11 a été utilisée comme approche pour le clonage moléculaire de l'ADN répétitif de Mycobacterium tuberculosis dont on a montré la présence sur un fragment génomique Alu I de 5,6 kb précédemment décrit. Le clone recombinant RI8.8.2, qui s'hybridisait fortement avec le fragment Alu de 5,7 kb marqué par radioactivité, contenait deux fragments Eco RI d'une taille de 2 kb et 1,4 kb. L'hybridisation de chacun de ces fragments à l'ADN de M. tuberculosis clivé par une endonucléasc de restriction a clairement démontré que le fragment de 2 kb contenait la séquence d'ADN répétitif, tandis qu'un seul exemplaire du fragment de 1,4 kb est représenté. Lorsque des répliques en provenance de la librairie génomique ont été sondées, le fragment génomique de 5,6 kb et l'insert répétitif cloné de 2 kb s'hybridisaient avec un numbre identique de plaques, indiquant la similarité et le grande nombre d'exemplaires de l'unité répétitive que les deux fragments avaient en commun. La cartographie par restriction et l'hybridisation indiquaient que le fragment répétitif d'ADN de 2 kb et l'exemplaire unique de 1,4 kb provenaient de parties contiguës de l'ADN génomique. Les deux fragments se révélèrent spécifiques aux membres du complexe M. tuberculosis, si ce n'est que le fragment de 2 kb montrait une hybridisation faible avec l'ADN de M. kansasii. Finalement, une région de 169 bp d'une extrémité de la séquence unique a été amplifiée par une réaction de polymerase en chaîne (PCR) d'une manière spécifique aux membres du complexe M. tuberculosis. La sensibilité de la PCR était telle que des bacilles en nombre aussi faible que 9 ou 10 pouvaient être détectés.RESUMEN
Se hizo el tamisaje de una genoteca preparada en λgt11 para después intentar la clonación de la secuencia repetitiva del DNA del Mycobacterium tuberculosis presente en el fragmento genómico Alu I de 5.6 kb. La clona recombinante R 18.8.2, la cual hibridizó fuertemente con el fragmento Alu 5.7-kb radiomarcado, contuvo dos insertos Eco RI de 2 kb y 1.4 kb, respectivamente. La hibridización de cada uno de estos fragmentos con el DNA del M. tuberculosis fraccionado por endonueleasas de restricción, demostró claramente que el fragmento de 2 kb contuvo la secuencia repetitiva de DNA, mientras que el framento de 1.4 kb se expresó en una sola copia. El análisis de la réplicas de las placas de la genoteca, mostró que el fragmento genómico de 5.6 kb y el inserto repetitivo de 2 kb, hibridizaron con un número idéntico de placas, indicando la similitud y el alto número de copias de la unidad repetitiva compartida por los 2 fragmentos. Los patrones de restricción y de hibridización indicaron que tanto la secuencia de DNA repetitiva (2 kb) como la contenida en una sola copia (1.4 kb), se originaron de secuencias contiguas en el DNA genómico. Se encontró que ambos fragmentos fueron exclusivos de los miembros del complejo del M. tuberculosis, aunque el inserto de 2 kb exhibió una débil hibridización con el DNA de M. kansasii. Finalmente, utilizando la reacción en cadena de la polimerasa (PCR) se ha amplificado una región de 169 pb de uno de los extremos déla secuencia contenida una sola copia (1.4 kb), la cual ha resultado ser especifica para los miembros del complejo M. tuberculosis. La sensibilidad del PCR fue tal que permitió detectar desde 9-10 bacilos.Tuberculosis continues to be a global health problem. Nearly one third of the world's population is reported to be infected with the causative agent Mycobacterium tuberculosis (19). In India alone there arc 9-10 million people suffering from the disease at any given time, with an annual mortality rate of 500,000 (13). The white plague has reappeared in the West as a result of the increase in the numbers of patients with AIDS. While several immunological studies are currently in progress to identify prophylactic/immunotherapeutic agents, an equally important task in combating the disease is to gain an understanding of the complex mycobacterial genome at the molecular level. There have been many reports of reiterated DNA sequences in M. tuberculosis (6,14,21). Nucleotide sequence data have shown some of these to be insertion elements (9,17). Hermans, et al, however, reported a 10-bp tandemly repeated DNA sequence which showed significant homology to the "chi" (crossover hotspot instigator) sequence of Escherichia coli (10). Characterization of repetitive elements and adjacently located genes may give insight into some of the molecular mechanisms that make mycobacteria so unique.
We have described earlier the identification of a 5.6-kb Alu fragment which is present in multiple copies in the genome of M. tuberculosis (14). We now report the a) molecular cloning and characterization of this repetitive element and a contiguously located single copy region of M. tuberculosis, and b) development of a highly specific and sensitive polymerase chain reaction (PCR)-based diagnostic assay for the detection of M. tuberculosis.
MATERIALS AND METHODS
Bacterial strains, phage library and plasmids. The bacterial strains listed in The Table were obtained from the standard collection of the Trudeau Institute, Saranac Lake, New York, U.S.A., except for M. tuberculosis south Indian isolate and Mycobacterium w, which are available at the National Institute of Immunology, New Delhi, India. The M. tuberculosis λgt11 genomic library was obtained from the World Health Organization. R18.8.2 is a phage clone which strongly hybridized to the 5.6-kb Alu fragment (14). The 2-kb and 1.4-kb Eco RI inserts of R18.8.2 were subcloned in bluescript plasmid vector, and the resulting clones were named pKP4 and pKSlO, respectively. Recombinant DNA techniques, such as extraction of genomic and plasmid DNA, restriction endonucleasc cleavage, purification of fragments from low melting point (LMP) agarose and Southern hybridization, were performed as described earlier (14).

Replica plaque lifts. A M. tuberculosis phage library was plated such that a 90-mm petri dish contained 103 plaque forming units (pfu). Genescreen nylon membrane (Dupont NEN, Boston, Massachusetts, U.S.A.) 88 mm in diameter was layered on top of the soft agarose and allowed to absorb phage for 60 sec. Then it was carefully peeled off the plate. A second membrane (the replica) was then placed on the same plate and allowed to sit for 90 sec before it was lifted off. The two membranes were processed for plaque hybridization as per Maniatis, et al. (11). The first membrane was probed with a 5.6-kb Alu fragment; the replica lift was probed with a 2-kb insert of pKP4.
Polymerase chain reaction (PCR). The oligonucleotide primers used for PCR-5'-CAACCAACATCGCGCTG-3' (KS3) and 5'-ATAGCCGATGACGGTCG-3' (K.S4)- were synthesized using a Gene Assembler (Pharmacia, Sweden). Typically, a 100-MI PCR reaction volume contained 1 x PCR buffer [10 raM Tris-HCl at pH 8.3, 50 mM KC1, 1.5 mM MgCl2, 0.01% (w/v) gelatin]; 200 µM of deoxynucleotide triphosphates (dNTP), 20 µM each of primers, 50 ng of genomic or 1 ng of plasmid DNAs and 2.5 units of Taq DNA polymerase. The mixture was overlaid with 100 µl of mineral oil. Each PCR cycle was performed at 94ºC for 1 min, 55ºC for 2 min, and 72ºC for 2 min; 30 such cycles were repeated in a thermal DNA cycler (Perkin-Elmer Cetus Instruments, Norwalk, Connecticut, U.S.A.). The amplified product was analyzed by electrophoresis on ethidium bromide containing 1.2% agarose gels, followed by Southern hybridization with the 169-bp product of pKSlO. For this purpose, the 169-bp fragment was eluted from LMP agarose gel and 32P-dCTP (alpha 32P-deoxycytidine triphosphate) labeled by primer extension using KS3 and KS4 primers. The filters in Figures 4B and 5B were hybridized in 5x Denhardt's [0.1% Ficoll (Pharmacia), 0.1% polyvinylpyrrolidone, and 0.1% bovine serum albumin (Fraction V; Sigma)] and 6x SSC (1 x SSC: 0.15 M sodium chloride, 0.015 M sodium citrate) at 65ºC overnight. Washings were done with 2 x SSC and 1% SDS (sodium dodecyl sulfate) at room temperature for 30 min, then at 65ºC for 1 hr and, finally, with 0.2 x SSC at 65ºC for 30 min. The specificity of the hybridization signal (as shown in Fig. 4B) was obtained after the wash with 2 x SSC at 65ºC for 1 hr.
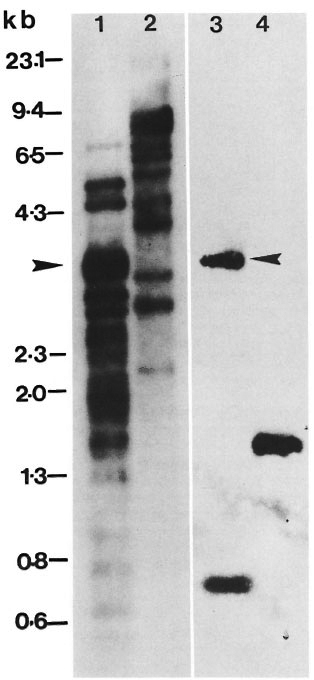
Fig. 1. Comparative Southern hybridization. Equal amounts (3 ng each) of M. tuberculosis H37Rv DNA digested to completion with Alu I (lanes 1, 3) or Eco RI (lanes 2, 4) were separated by 1.2% agarose gel electrophoresis, Southern transferred and hybridized to "P-dCTP labeled 2-kb (lanes 1, 2) or 1.4-kb (lanes 3, 4) inserts of pKP4 and pKSlO, respectively. Arrow = position of 3.5-kb Alu fragment in lanes 1 and 3 to which both probes are seen to hybridize; molecular size markers in kilobase pairs are indicated on the left.
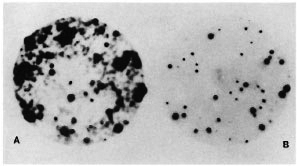
Fig. 2. Copy number and similarity of the repeating unit shared by the 5.6-kb Alu I genomic fragment and 2-kb cloned insert. M. tuberculosis phage library was plated at 103 pfu per 90-mm petri dish, replica plaque lifts were probed with either 5.6-kb Alu fragment (A) or 2-kb cloned insert (B). A total of 1018 plaques were present on this petri dish. Of the 40% recombinants, 48 hybridized with cither probe. Plaque transfer was more efficient in A; hence the intensity of the signal as compared to the second plaque lift in panel B. Striking similarity in hybridization signals is noticeable in both A and B.
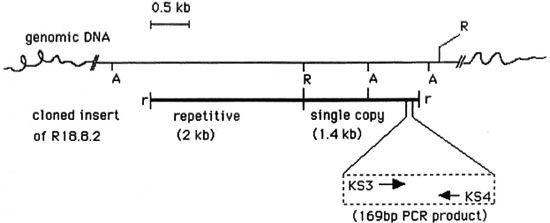
Fig. 3. Repetitive (2-kb) and single copy (1.4-kb) sequences arc contiguously located in the M. tuberculosis genome. Physical maps of complete insert of R 18.8.2 (thick bar) and chromosomal region ( --- bar) from where cloned insert originated. Parts of the chromosomal DNA between Alu I sites correspond to 3.5-kb and 0.8-kb Alu bands seen in Fig. 1, lane 3. Location and direction of synthesis of PCR primers KS3 and KS4 is also depicted. A = Alu I, R = Eco RI, r = linked Eco RI. There were single sites for Sac II, Bst EII and Sma I on the 2-kb insert. The following sites were not present on the 2-kb repetitive DNA: Alu I, Kpn I, Xho I, Acc I, Sal I, Cla I, Hind III, Pst I, Bam HI and Sau 3A.
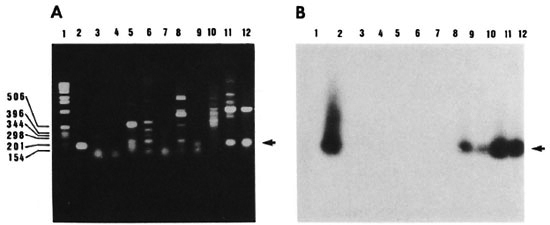
Fig. 4. Specificity of PCR . Agarose gel electrophoresis of amplified DNAs of different mycobacterial strains using oligonucleotide pair KS3 and KS4 (A) and autoradiograph of the transferred DNA hybridized to the radiolabeled 169-bp product of pKSlO (B). Lanes 1 = 1-kb size marker; 2 = plasmid clone pKSlO; 3 = M. vaccae; 4 = M. smegmatis; 5 = M. kansasii; 6 = M. scrofulaceum; 1 = M. intracellulars, 8 = M. avium; 9 = M. bovis BCG; 10 = M. tuberculosis south Indian isolate; 11 = M. tuberculosis H37Rv; 12 = M. tuberculosis H37Ra. Arrow = position of the 169-bp band; molecular size markers are listed in basepairs.
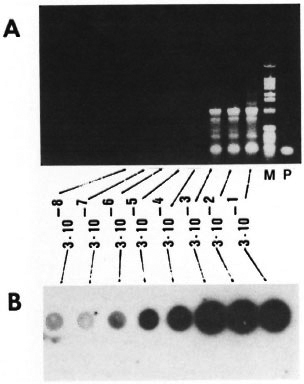
Fig. 5. Level of detection by PCR amplification. Tenfold serial dilutions ofM tuberculosis genomic DNA ranging from 300 ng to 30 fg (3 x 10-1 to 3 x 10-8 µg) were PCR amplified using KS3 and KS4 primers. One tenth volume of PCR products were electrophorcscd on 1.2% agarose gels (A) or dotted and hybridized with radiolabeled 169-bp probe (B). Lane M = 1-kb ladder as idicatcd in Fig. 4. Lane P has the 169-bp PCR amplification product of pKSlO.
RESULTS
Subcloning. In order to identify recombinant clones containing repetitive DNA, a λgt11 genomic library of M. tuberculosis H37Rv was screened with the 5.6-kb Alu fragment (14). One of the strongly hybridizing phage clones, R 18.8.2, was plaque purified and characterized. Eco RI digestion of R 18.8.2 DNA released two insert fragments of 2-kb and 1.4-kb that were subcloned in bluescript plasmid vectors. The subclones have been referred to as pKP4 and pKSlO, respectively. Comparative Southern hybridization. A comparative Southern hybridization experiment was designed to determine whether one or both inserts of R 18.8.2 possess a repetitive element. Equal amounts of Alu Iand Eco Rl-digested M. tuberculosis DNA were run in duplicate in parallel lanes (see legend to Fig. 1). Following Southern transfer, lanes 1 and 2 were hybridized with the pKP4 insert and lanes 3 and 4 with the pKSlO insert. Southern hybridization results have shown the pKP4 insert to be hybridizing to multiple DNA fragments in the Alu I digest and to several high molecular size bands in the Eco RI digest (Fig. 1, lanes 1, 2). On the other hand, the pKSlO insert hybridized to two bands in the Alu I digest and to one band in the Eco RI digest (Fig. 1, lanes 3, 4). The hybridization pattern of the pKP4 insert resembled that of the previously reported 5.6-kb Alu fragment (14) in that it hybridized to multiple DNA fragments of similar size; whereas that of the pKSlO insert was of a type consistent with single copy genes. The results of this experiment, therefore, suggested that M. tuberculosis repetitive DNA sequence, homologous to the genomic 5.6-kb Alu fragment, is contained within the cloned 2-kb insert of pKP4.
Comparative plaque hybridization. The Southern hybridization of the pKP4 insert to multiple bands in the genomic digests indicated the high copy number of the repeat. In order to examine the extent of representation of this repeat in the genome of M. tuberculosis and to further confirm the similarity of the nature of the repetitive element snared by the 5.6-kb Alu fragment and the cloned 2-kb insert, a comparative plaque hybridization was performed. A M. tuberculosis phage library was plated at a density of 103 pfu per 90-mm petri plate. Replica plaque lifts were then probed with either the pKP4 insert or the 5.6-kb Alu fragment. As expected, the plaque hybridization signals obtained with the two probes were identical. Typical results are shown in Figure 2. Out of the 1018 plaques on this plate, of which only 40% were recombinant, as many as 48 strong super imposable hybridization signals were observed with either probe, indicating that nearly 10% of the recombinant clones in the M. tuberculosis genomic library contained the repetitive DNA.
2-kb Repetitive and 1.4-kb single copy inserts of R18.8.2 are contiguously located in M. tuberculosis genome. The λgt11 genomic library used in this study was constructed by adding Eco RI linkers to sheared, end repaired and Eco RI methylase protected genomic DNA (20). Two possibilities therefore existed: 1) The two Eco RI fragments of R18.8.2 (2 and 1.4 kb) could have originated from different regions of the genome but got ligated together because of Eco RI linkers, or 2) the entire 3.4-kb insert of R 18.8.2 is one contiguous piece of genomic DNA and an internal Eco RI site cleaved it into the 2-kb repetitive and 1.4-kb single copy fragments. That the latter is true is indicated by the hybridization of 2-kb and 1.4-kb inserts to a common 3.5-kb Alu fragment in the genomic DNA digest of M. tuberculosis (Fig. 1, arrow). A partial restriction map showed that there are no Alu I sites on the pKP4 insert and one Alu site is present on the pKSlO insert. By virtue of the absence of the Alu I site, the pKP4 insert hybridized to the larger 3.5-kb genomic Alu fragment from which it originated. The pKSlO insert, which had an internal Alu I site, also hybridized with this 3.5-kb band (of which it forms a part) and with an additional genomic band of 0.8 kb (Fig. 1, lane 3). The hybridization of the pKP4 and pKSlO insert DNAs to the common 3.5-kb genomic Alu fragment, therefore, proved that the repetitive element (pKP4 insert) and the single copy region (pKSlO insert) originated from this contiguous piece of genomic DNA, as has been schematically represented in Figure 3.
Diagnostic potential. To evaluate the usefulness of the cloned inserts in the diagnosis of M. tuberculosis, the specificity of hybridization was determined for pKP4 and pKS 10. Two µM each of genornic DNAs from M. tuberculosis and several other mycobacteria were dotted in duplicate and hybridized with radiolabeled probes. The repetitive pKP4 insert hybridized strongly with M tuberculosis strains, M. bovis BCG and M. africanum DNA, but not with M. avium, M. intracellulare, M. scrofulaceum, M. smegmatis, M. cheloni, M. fortuitum, M. vaccae or Mycobacterium w. However, there was a weak crosshybridization with M. kansasii DNA. On the other hand, the single copy pKSlO insert appeared to be specific in its hybridization with members of the M. tuberculosis complex. There was no reactivity with any of the other mycobacteria, including M. kansasii. These results are summarized in The Table. Neither cloned fragment showed hybridization with genomic DNA from a number of nonmycobacterial organisms or human DNA (data not shown).
PCR-based diagnostic assay. The specificity of the 1.4-kb single copy insert to the M. tuberculosis complex suggested that a target region of appropriate length could be chosen from this sequence for PCR amplification (16). The pKSlO insert has been completely sequenced (Reddi, et al., unpublished results). Oligonucleotide primers KS3 and KS4 were designed such that a 169-bp band could be amplified from the genome of M. tuberculosis as shown in Figure 3, lower panel.
Specificity of the PCR amplification. Genomic DNAs from members of the M. tuberculosis complex as well as from other mycobacteria (see legend to Fig. 4) were PCR amplified using the KS3 + KS4 primer combination. The expected 169-bp fragment was amplified in M. tuberculosis H37Ra, M. tuberculosis H37Rv and M. bovis BCG (Fig. 4 A, lanes 9, 11, 12, arrow) which was of the same size as the amplified product of plasmid clone pKSlO, used as a positive control (Fig. 4A, lane 2). DNAs from other strains of mycobacteria were also amplified (Fig. 4A, lanes 3-8). However, the sizes of the amplified fragments were different from those obtained with M. tuberculosis DNA. In order to differentiate the M. tuberculosis complex-specific PCR amplified product from the rest, the 169-bp PCR product of pKSlO was radiolabeled and used as probe to hybridize to the gel in Figure 4A. On Southern hybridization, a 169-bp hybridization signal was seen only in lanes containing M. tuberculosis H37Rv, M. tuberculosis H37Ra, M. tuberculosis south Indian isolate and M. bovis BCG DNAs (Fig. 4B, lanes 9-12). Even though several amplified fragments were visualized on ethidium-bromide-stained gel, there was no hybridization with any of the other mycobacteria tested (Fig. 4B, lanes 3-8) thereby putting in evidence the specificity of PCR. It is also of relevance that Southern hybridization revealed the presence of 169-bp amplified product in the Indian isolate of M. tuberculosis (Fig. 4B, lane 10). The target sequence chosen for PCR amplification with amplimers KS3 and KS4 appeared to be conserved among strains of M. tuberculosis, thereby indicating that the PCR test is suitable for application to clinical specimens.
Sensitivity of PCR. The sensitivity of the PCR with primers KS3 and KS4 was determined by adding to the reaction mixtures serial dilutions of purified M. tuberculosis DNA ranging from 300 ng to 30 fg per assay. Up to 300 pg of chromosomal DNA, the amplified product could be detected by agarose gel electrophoresis followed by ethidium-bromide staining (Fig. 5A); whereas the amplified product from 30 fg of chromosomal DNA could be detected by a simple dot hybridization using a radiolabeled 169bp PCR product as probe (Fig. 5B). Given the size of the M. tuberculosis genome as 2.5 x 109 Da, 30 fg of DNA corresponds to approximately 9-10 bacilli. This experiment demonstrated that with PCR amplification followed by hybridization, one could detect as few as 9-10 tubercle bacilli. In order to examine the factors that could inhibit the PCR, unrelated DNA from either M. smegmatis or human leukocytes was added to simulate the presence of extraneous DNA in the PCR reaction mixtures. This did not affect the level of sensitivity of the assay (data not shown). To rule out PCR contamination and false-positive amplification, we routinely included a tube with no template DNA and constantly found this to be negative for amplification (data not shown).
DISCUSSION
Screening of a M. tuberculosis genomic library with the 5.6-kb Alu fragment led to the identification of a clone bearing 2-kb and 1.4-kb inserts. Hybridization to M. tuberculosis genomic DNA digests showed that the 2-kb fragment (pKP4) hybridized to multiple bands of sizes ranging from 6.5 to 0.6-kb. This indicated the presence of a repetitive sequence within this cloned fragment. The 1.4-kb fragment (pKSlO), on the other hand, has shown a hybridization pattern which is typical of single copy genes. It is to be noted that the 2-kb probe hybridized intensely with the 5.6-kb Alu fragment. Further evidence that the cloned pKP4 and the originally identified 5.6-kb genomic Alu fragment contain essentially the same repetitive element came from the plaque hybridization experiment. Replica plaque lifts of the M. tuberculosis library gave identical hybridization signals when probed with either the 5.6-kb Alu fragment or the cloned 2-kb probe. The ratio of plaques hybridized to the total number of recombinant plaques plated gave an indication that the repetitive sequence is found on 10% of the cloned genomic fragments of the M. tuberculosis library.
There have been several reports of wellcharacterized, repetitive DNA sequences of M. tuberculosis (9,17). IS 6110 and IS 986 have been shown to be mobile elements. Both these sequences contain several Alu I sites; whereas the pKP4 repeat does not contain any Alu I site and is, therefore, not the same as IS67 10 or IS986. Hermans, et al. (8) have reported cloning of the M. tuberculosis repetitive fragment pPH7301 based on the reactivity with monoclonal antibody F116-5. The nucleotide sequence of pPH7301 showed the presence of 10-bp repeats that were termed as major polymorphic tandem repeats (MPTR) (10). Amplimer pairs A and D or I and F (8,10) which amplify 158- and 181 -bp fragments of pPH7301, respectively, failed to amplify pKP4 DNA (data not shown). Further, the presence of two Sau 3A sites in pPH7301 and none in pKP4 proved that these two repeated sequences are not the same. Ross, et al. have reported the cloning of high molecular weight Nde II fragments of M. tuberculosis which contain repetitive DNA (15). These investigators appeared to have cloned essentially the same repetitive element as is present on the 5.6kb Alu fragment previously reported by our group (14). This is evident from the Southern hybridization profiles of pTBN12 and pTBN52 to Alu I-digested M. tuberculosis DNA (15) which are identical to that of the 5.6-kb Alu fragment (14) and the present pKP4 repeat. Interestingly enough, the restriction maps of the three clones, pKP4, pTBN12 and pTBN52, are distinctly different. We believe that this argues against the presence of a coding region in these repeats. It could be conceived then that the actual repetitive unit, which is likely to be common to pKP4, pTBN12 and pTBN52, is small in size and is present at multiple loci in the genome of M. tuberculosis. By employing different routes of molecular cloning, our group and Ross, et al. have identified genomic fragments originating from different loci but yet containing the same repetitive unit. While these repeated elements have proven to be useful epidemiological markers (3,10,15,18), the functional significance of the presence of such presumably noncoding repeats at multiple loci in the genome remains to be elucidated. It is also in this context that the characterization of contiguously located gene sequences, such as the 1.4 kb of the present study, becomes relevant. Grossinsky, et al. (7) reported the location of a species-specific repetitive DNA downstream of the coding region of the 65-kDa antigen gene in M. leprae. We arc further investigating if the pKP4 M. tuberculosis repeat is located near a particular class of genes by characterizing additional clones which contained the repeat sequence.
Conventional methods used for the diagnosis of tuberculosis infection have inherent limitations. Bacteriological examination by microscopy is simple, rapid and economical, but is nonspecific and can detect only the heavily infected cases. The culture technique, on the other hand, is highly sensitive and specific but it takes 6-8 weeks. DNA probe technology combines the speed of microscopy with the reliability and accuracy of the culture technique and should, therefore, be considered as an alternative method for the diagnosis of tuberculosis.
Diagnostic potential has been assessed for the cloned M. tuberculosis DNA sequences pKP4 and pKSlO identified in the present study. The repetitive as well as the single copy region were specific in hybridization to members of the M. tuberculosis complex, except that the former exhibited weak crosshybridization with M. kansasii. In this regard, pKSlO showed the specificity desired of a diagnostic probe. The reported detection limit of a radiolabeled DNA probe is approximately 10,000 bacilli per assay (12). In most clinical situations, especially in extra pulmonary tuberculosis and paucibacillary cases, the bacilli in a test sample tend to be fewer than 1000 which cannot be picked up by the direct probe assay. In order to be able to detect M. tuberculosis directly in a clinical specimen without the need for culture, we have developed a PCRbased diagnostic assay.
PCR amplification of a defined target sequence and hybridization with a specific probe is a highly sensitive and reliable method for the early detection of M. tuberculosis. We have chosen a 169-bp region from the 1.4-kb single copy fragment as a target for PCR amplification by amplimers KS3 and KS4. By PCR amplification followed by hybridization with the radiolabeled 169-bp PCR product of pKSlO, we were able to demonstrate that 1) the assay is highly specific to M. tuberculosis and M. bovis and 2) the sensitivity of the assay has improved from roughly 1000 bacilli detectable by ethidium-bromide-stained gel to as few as 9-10 bacilli.
Several reports have appeared describing the PCR assay for the detection of M. tuberculosis (1,2,4,5,8). These differ in the target region chosen for amplification. Although the PCR technique offers a high degree of sensitivity, care should be taken to eliminate false-negative and false-positive results. Some investigators read the PCR results from ethidium-bromide-stained agarose gels alone (2); others introduced an additional confirmatory step either in the form of restriction endonuclease digestion of PCR product (4) or hybridization with an oligonucleotide probe internal to the amplified fragment (1). However, impurities often inhibit cleavage by restriction endonucleases which makes interpretation of the results difficult. It should also be borne in mind that Taq polymerase, which lacks proofreading ability, may introduce mismatched bases into the amplified product which, theoretically, would alter a restriction enzyme site or the nucleotide sequence to be hybridized with the internal oligo probe. We believe that our method of using the radiolabeled, whole 169-bp probe for hybridization to the PCR products not only safeguards against these possible artifacts but offers the additional advantage of higher sensitivity. Conversion of the labeling of the 169-bp probe to a nonisotopic modality and analysis of a large number of clinical samples from proven and suspected cases of tuberculosis to evaluate the utility of our PCR diagnostic assay is currently in progress.
Acknowledgment. We thank Ashis Das (NII) for the synthesis of PCR primers and Dr. Rama Mukherjee (NII) for providing us with mycobacterial strains.
REFERENCES
1. BRISSON-NOEL, A., GICQUEL, B., LECOSSIER, D., LEVY-FREBAULT, V., NASSIF, X. and HANCE, A. J. Rapid diagnosis of tuberculosis by amplification of mycobacterial DNA in clinical samples. Lancet 2(1989)1069-1071.
2. BUCK, G. E., O'HARA, L. C. and SUMMERSGILL, J. T. Rapid, simple method for treating clinical specimens containing Mycobacterium tuberculosis to remove DNA for polymerase chain reaction. J. Clin. Microbiol. 30(1992)1331-1334.
3. CAVE, M. D., EISENACH, K. D., MCDERMOTT, P. F., BATES, J. H. and CRAWFORD, J. T. IS6110: conservation of sequence in Mycobacterium tuberculosis complex and its utilization in DNA fingerprinting. Mol. Cell. Probes 5(1991)73-80.
4. COUSINS, D. V., WILTON, S. D., FRANCIS, B. R. and Gow, B. L. Use of polymerase chain reaction for rapid diagnosis of tuberculosis. J. Clin. Microbiol. 30(1992)255-258.
5. DE WIT, D., STEYN, L., SHOEMAKER, S. and SCOGIN, M. Direct detection of Mycobacterium tuberculosis in clinical specimens by DNA amplification. J. Clin. Microbiol. 28(1990)2437-2441.
6. EISENACH, K. D., CRAWFORD, J. T. and BATES, J. H. Repetitive DNA sequences as probes for Mycobacterium tuberculosis. J. Clin. Microbiol. 26(1988)2240-2245.
7. GROSSINSKY, C. M., JACOBS, W. R., CLARK-CURTISS, J. E. and BLOOM, B. R. Genetic relationships among Mycobacterium leprae, Mycobacterium tuberculosis, and candidate leprosy vaccine strains determined by DNA hybridization: identification of an M. leprae -specific repetitive sequence. Infect. Immun. 57(1989)1535-1541.
8. HERMANS, P. W. M., SCHUITEMA, A. R. J., VAN SOOLINGEN, D., VERSTYNEN, C. P. H. J., BIK, E. M., THOLE, J. E. R., KOLK, A. H. K. and VAN EMBDEN, J. D. A. Specific detection of Mycobacterium tuberculosis complex strains by polymerase chain reaction. J. Clin. Microbiol. 28(1990)1204-1213.
9. HERMANS, P. W. M., VAN SOOLINGEN, D., DALE, J. W., SCHUITEMA, A. R. J., MCADAM, R. A., CATTY, D. and VAN EMBDEN, J. D. A. Insertion element IS986 from Mycobacterium tuberculosis: a useful tool for diagnosis and epidemiology of tuberculosis. J. Clin. Microbiol. 28(1990)2051-2058.
10. HERMANS, P. W. M., VAN SOOLINGEN, D. and VAN EMBDEN, J. D. A. Characterization of a major polymorphic tandem repeat in Mycobacterium tuberculosis and its potential use in epidemiology of Mycobacterium kansasii and Mycobacterium gordonae. J. Clin. Microbiol. 12(1992)4157-4165.
11. MANIATIS, T., FRITSCH, E. F. and SAMBROOK, J. Molecular Cloning: A Laboratory Manual. Cold Spring Harbor, New York: Cold Spring Harbor Laboratory, 1982.
12. PAO, C. C, LYN, S. S., WU, S. Y. and JUANG, W. M. The detection of mycobacterial DNA sequences in uncultured clinical specimens with cloned Mycobacterium tuberculosis DNA as probes. Tubercle 69(1988)27-36.
13. PARK, J. E. and PARK, K. Textbook of Preventive and Social Medicine. Jabalpur, India: M/ S Banarsidas Bhanot, 1986, pp. 164-177.
14. REDDI, P. P., TALWAR, G. P. and KHANDEKAR, P. S. Repetitive DNA sequence of Mycobacterium tuberculosis: analysis of differential hybridization pattern with other mycobacteria. Int. J. Lepr. 56(1988)592-597.
15. Ross, B. C, RAIOS, K., JACKSON, K. and DWYER, B. Molecular cloning of a highly repeated DNA element from Mycobacterium tuberculosis and its use as an epidemiological tool. J. Clin. Microbiol. 30(1992)942-946.
16. SAIKI, R. K., GELFAND, D. H., STOFFEL, S., SCHARF, S. J., HIGUCHI, R., HORN, G. T., MULLIS, K. B. and ERLICH, H. A. Primer directed enzymatic amplification of DNA with a thermostable DNA polymerase. Science 239(1988)487-491.
17. THIERRY, D., CAVE, M. D., EISENACH, K. D., CRAWFORD, J. T., BATES, J. H., GICQUEL, B. and GUESDON, J. L. IS6110, an IS-like clement of Mycobacterium tuberculosis complex. Nucleic Acids Res. 18(1990)188.
18. VAN SOOLINGEN, D., HERMANS, P. W. M., DE HAAS, P. E. W., SOLL, D. R. and VAN EMBDEN, J. D. A. Occurrence and stability of insertion sequences in Mycobacterium tuberculosis complex strains: evaluation of an insertion sequence-dependent DNA polymorphism as a tool in epidemiology of tuberculosis. J. Clin. Microbiol. 29(1991)2578-2586.
19. WORLD HEALTH ORGANIZATION. Progress and evaluation report, Tuberculosis Control Programme, EB87/4. Geneva: World Health Organization, November 1990.
20. YOUNG, R. A., BLOOM, B. R., GROSSINSKY, C. M., IVANYI, J., THOMAS, D. and DAVIS, R. W. Dissection of Mycobacterium tuberculosis antigens using recombinant DNA. Proc. Natl. Acad. Sci. U.S.A. 82(1985)2583-2587.
21. ZAINUDDIN, Z. F. and DALE, J. W. Polymorphic repetitive DNA sequences in Mycobacterium tuberculosis detected with a gene probe from Mycobacterium fortuitum plasmid. J. Gen. Microbiol. 135(1989)2347-2355.
1. Ph.D.; National Institute of Immunology, New Delhi 110067, India.
2. D.Sc; National Institute of Immunology, New Delhi 110067, India.
3. Ph.D., National Institute of Immunology, New Delhi 110067, India.
Present address for Dr. Reddi: Department of Anatomy and Cell Biology, Box 439, School of Medicine, University of Virginia, Charlottesville, Virginia 22908, U.S.A.
Reprint requests to Dr. Khandekar.
Received for publication on 11 March 1991.
Accepted for publication in revised form on 22 January 1993.