- Volume 61 , Number 2
- Page: 236–44
Serologic responses to nerve antigens in sooty mangabey monkeys with experimental leprosy
ABSTRACT
Eight sooty mangabey monkeys were inoculated intravenously and intradermally with varying doses of Mycobacterium leprae f rom 4.8 x 107 to 4.8 x 1010. Serum samples were obtained f rom the animals at intervals of about 3 months for 90 months, and were examined for IgM and IgG antibodies to nerve antigens, including ceramide, galactocerebroside (GC), and asialo-GM, (AGM1), using an enzyme-linked immunosorbent assay (ELISA). The serological results were then compared with clinical findings, particularly nerve involvement. Of 8 mangabey monkeys inoculated with M. leprae, 7 animals had clinical leprosy; 6 of them had nerve damage, including neurologic deformities in 4 monkeys and nerve enlargement in 2. Median time for the initial signs of leprosy was 10 months postinoculation (p.i.), a range f rom 4 to 35 months. In contrast, nerve damage was noted rather late, about 35 to 86 months p.i. (median 54 months). The major immunoglobulin class to ceramide, GC, and AGM1 antigens was IgM, and the antibody responses to the nerve antigens appeared f rom 15 to 63 months p.i. (median 37 months). Antineural antibodies were thus detectable about 18 months (range -2 to 60 months) prior to observable nerve damage. In addition, elevation of antineural antibody levels were predictive of clinical exacerbation of the disease and neuritic damage. This study suggests that antineural antibodies are produced during the course of M. leprae infection and may be indicative of nerve damage, such as neurological deformities or nerve enlargement, in leprosy patients.RÉSUMÉ
On a inoculé huit singes mangabey par voies intraveineuse et intradermique avec du Mycobacterium leprae à des doses variant de 4, 8 x 107 à 4, 8 X 1010. On a prélevé des échantillons de serum chez les animaux à intervalles d'environ trois mois durant nonante mois, et on a recherché la présence d'anticorps IgG et IgM vis-à-vis d'antigènes nerveux, y compris le ceramide, legalactocebroside (GC) et l'asialo-GM, (AGM1) par un test immuno-enzymatique (ELISA). Les résultats sérologiques ont été comparés avec les observations cliniques, en particulier l'atteinte nerveuse. Parmi les huit singes mangabey inoculés avec du M. leprae, sept avaient une lèpre clinique; six d'entre eux avaient une atteinte nerveuse, avec des déformités d'origine neurologique chez quatre singes et un épaississemente de nerfs chez deux d'entre eux. L'intervalle de temps médian pour l'apparition des premiers signes de lèpre suite à l'inoculation était de 10 mois, allant de 4 à 35 mois. Par contre, l'atteinte nerveuse a été notée plutôt tardivement, environ 35 à 86 mois après l'inoculation (médiane 54 mois). La principale classe d'immunoglobulines vis-à-vis des antigènes ceramide, GC, et AGM1 était du type IgM et les réponses par anticorps aux antigènes nerveux sont apparus entre le quinzième et le soixante-troisième mois après l'inoculation (médiane 37 mois). Des anticorps dirigés contre les nerfs étaient donc détectables environ 18 mois (de 2 à 60 mois) avant qu'on puisse observer des dégâts nerveux. De plus, une augmentation des taux d'anticorps vis-à vis des nerfs prédisait une exacerbation clinique de la maladie et une détérioration nerveuse. Cette étude suggère que des anticorps vis-àvis des nerfs sont produits dans le décours de l'infection par M. leprae et peuvent indiquer une atteinte nerveuse chez les malades de la lèpre, telle que des déformités d'origine neurologique ou un épaississement des nerfs.RESUMEN
Se inocularon 8 monos mangabey pardos con difcrentres cantidades de Mycobacterium leprae (de 4.8 x 107a 4.8 X 1010) por las vías intravenosa c itradermiea. Después, a intervalos de 3 meses, se obtuvieron muestras de suero durante 90 meses, y en los sueros se buscaron anticuerpos I gM c IgG contra antígenos del sistema nervioso, incluyendo un cerámido, un galactoccrebrósido (GC), y un asialo-GM, (AGM1), usando un inmunoensayo enzimático (ELISA). Los resultados scrológicos se compararon con los hallazgos clínicos, particularmente con el daño a nervios. De los 8 monos mangabey inoculados con M. leprae, 1 animales desarrollaron la enfermedad: 6 de ellos mostraron daño a nervios, incluyendo deformidades ncurológicas en 4 monos y engrosamiento nervioso en 2 de ellos. El tiempo promedio de aparición de los síntomas de la lepra fue de 10 meses postinoculación (p.i.), con un rango de 4 a 35 meses. En contraste, el daño a nervios se notó más tardiamente, entre los 35 y los 86 meses p.i. (mediana d3 54 meses). La inmunoglobulina predominante contra los tres antígenos (cerámido, GC y AGM1) fue de la clase IgM, y las respuestas en anticuerpos contra los antigenos aparecieron entre los 15 y los 63 meses p.i. (mediana de 37 meses). Así, los anticuerpos antineurales fueron detectados aproximadamente 18 meses (rango de -2 a 60 meses) antes de la aparición clínica del daño a nervios. Además, la elevación de los niveles de anticuerpos antineurales fue predictiva de la exacerbación clínica de la enfermedad y del daño neurítico. Este estudio sugiere que los anticuerpos antineurales se producen durante el curso de la infección por el M. leprae y que su presencia puede ser indicativa del daño a nervios. Esto podría ocurrir en los pacientes con lepra que muestran alteraciones ncurológicas o engrosamicnto de nervios.Nerve damage has been noted, with varying degrees of involvement, in virtually all leprosy patients. Although the pathogenic mechanisms of nerve destruction are not fully understood, antibodies to various nerve antigens have been implicated in the patho genesis of nerve damage in leprosy (22,27) However, whether or not antineural antibodies are significantly elevated in sera from leprosy patients is controversial (2,5,14,23,24,27) addition to technical differences between studies, this may be due partly to the fact that leprosy patients are diagnosed at different stages of the Mycobacterium leprae infection and have wide variations in nerve damage (3,11,18). Thus, it may be almost impossible to predict consistent relationships between antineural antibodies and the degree of nerve involvement in leprosy patients.
As a model of human leprosy, experimental animals such as armadillos and monkeys have been explored (13,26). Sooty mangabey monkeys are reported to be an excellent model for leprosy, with clinical signs and humoral responses resembling human leprosy, including neurological deformities (6). In this study, serial serum samples, taken at regular intervals from mangabey monkeys before and after inoculation with M. leprae, were examined for antibodies to nerve tissue antigens such as ceramide, galactocerebroside (GC), and asialo-GM, (AGM1). The antineural antibodies were then compared with clinical findings, including nerve involvement before and after chemotherapy. In almost every animal, antibodies to one or more of the nerve antigens were elevated significantly 1 to 2 years prior to neurological deformities. In this study we present convincing evidence that antineural antibodies are produced during the course of M. leprae infection and are associated with clinical status and/or the development of subsequent nerve damage.
MATERIALS AND METHODS
Animals and M. leprae inoculation. Eight sooty mangabey monkeys (Cercocebus torquatus atys) were divided into four groups, two animals in each group. Each pair of mangabeys was inoculated intravenously and intracutaneously with a total dose of M. leprae ranging from 4.8 x 107 to 4.8 x 1010 (morphological index 10%) in one-log increments between groups. The biological history of mangabeys, the preparation of M. leprae, inoculation procedures, and clinical evaluations have been described in detail previously (6). When the animals needed treatment, rifampin was given at approximately 10 mg/kg daily.
Serum specimens. Serum specimens were obtained from mangabeys 2 months before inoculation and at intervals of approximately 3 months postinoculation (p.i.). Serum samples were stored frozen until used for detecting antineural antibodies.
Nerve antigens. Ceramide and GC antigens were prepared from formalinized human peripheral nerve tissues. After soaking the formalinized nerve tissue in distilled water for 3 days with several changes, the tissues were freeze-dried. Lipids were then extracted with a chloroform : methanol (2:1) solution. After washing with a chloroform : methanol: water (4:2:1) solution, the lipids were applied to a silica gel (70 to approximately 230 mesh; Aldrich Chemical Co., Inc., Milwaukee, Wisconsin, U.S.A.) column and eluted with 2 bed volumes of chloroform, followed by 5%, 10%, and 20% (v/v) methanol in chloroform, respectively. Ceramide was identified from the lipid fraction eluted with 10% methanol and GC from the 20% methanol fraction, respectively, by comparing with standard ceramide and GC (Sigma Chemical Co., St. Louis, Missouri, U.S.A.) in thin-layer chromatography (TLC). Ceramide and GC antigens were then purified by preparative TLC. AGM1 was obtained commercially from Sigma.
Detection of antibodies to nerve antigens. Antibodies to nerve antigens were detected using an enzyme-linked immunosorbent assay (ELISA) (25) with minor modifications as previously described (4). Briefly, the nerve antigens were dissolved in ethanol at a 5.0 µg/ml concentration. A volume of 50 µl of each antigen was added to wells of a 96wcll, U-bottom microtiter plate (Dynatech Laboratories, Inc., Alexandria, Virginia, U.S.A.), and incubated at room temperature until completely evaporated. The wells were then washed with phosphate buffered saline (PBS), pH 7.4, and blocked by the addition of 200 µl of PBS-0.5% (w/v) BSA (ICN ImmunoBiologicals, Lisle, Illinois, U.S.A.). After emptying the wells, 50 µl of serum diluted 1:200 in PBS-5% normal goat serum (NGS) (Gibco Laboratories, Grand Island, New York, U.S.A.) was added to the wells and incubated at 37ºC for 90 min. This was followed by the addition of peroxidaseconjugated goat antihuman IgG or IgM (Cappel, Organontecknika Corp., West Chester, Pennsylvania, U.S.A.) diluted 1:3000 in PBS-5% NGS and of substrate solution, H2O2 + O -phenylenediamine. The absorbance was read at 490 nm.
Each serum was tested against all three antigens in the same plate. A pooled positive control serum was examined in each plate, and minor variations in the absorbances were corrected based on positive control values. Each test was performed in duplicate, and the absorbance of the wells without antigens was subtracted from those with the antigens before analysis.
RESULTS
Following inoculation of mangabey monkeys with M. leprae, serial serum specimens were obtained and examined for antibodies to nerve antigens such as ceramide, GC, and AGM1. The results were then compared with the clinical findings, especially for nerve involvement. Although both IgG and IgM antibodies to the antigens were investigated, only the ones which were elevated are shown in the figures. In general, there was no correlation between the inoculation doses of M. leprae and the antibody responses to the nerve antigens. Even between animals inoculated with the same doses of M. leprae, no consistent antibody responses to the nerve antigens were noted.
Mangabey D171, inoculated with 4.8 x 1010 M. leprae, developed lepromatous (LL) type leprosy 5 months postinoculation (p.i.) and was treated at 38 months (6). In this mangabey, small peaks of IgG antibodies to ceramide and GC antigens appeared at 6 and 3 months p.i., respectively (Fig. 1A). The first major IgM antibody peaks appeared at 15 months, followed by the second major IgM antibody responses between 30 and 44 months p.i. After chemotherapy was initiated at 38 months, IgM antibodies to ceramide and GC remained at a high level for an additional 6 months and then declined sharply by 54 months p.i. Meanwhile, major peaks of IgG antibodies to the ceramide and GC antigens were shown at 21 months and were followed by another minor IgG peak at 42 months p.i. A small peak of IgM antibodies to AGM1 was noted at 24 months only, and there were no significant IgG antibodies to the AGM1 antigen noticeable throughout the study period. Despite early strong antineural antibody responses and clinical leprosy by 15 months, neurologic deformities appeared obvious only at 75 months and persisted beyond 105 months p.i. Therefore, antineural antibodies were detectable 60 months earlier than neurologic deformities in this animal.
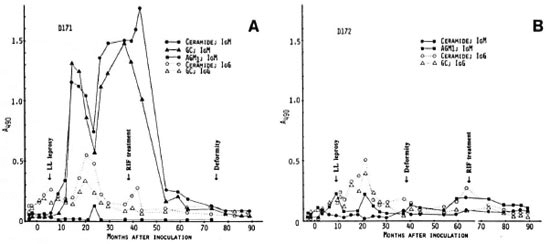
Fig. 1. Longitudinal antibody responses to nerve antigens in mangabeys D171 (A) and D172 (B) before andat intervals after inoculation with 4.8 x 1010 M. leprae.
In mangabey D172, also inoculated with 4.8 x 1010 M. leprae, clinical signs of disseminated lepromatous (LL) leprosy were found about 9 months p.i. and regressed spontaneously, but the disease recurred and regressed several times during the study period (6). Although antibody responses to the nerve antigens were not strong, there were several IgM antibody peaks to AGM1 (Fig. 1B). Interestingly, the major antibodies to ceramide and GC were of the IgG class; no significant IgM antibodies to the antigens were noted in mangabey D172. Neurologic damage was first noted at 39 months p.i. and became progressively worse beyond 90 months p.i. even though chemotherapy was received at 65 months p.i.; neurologic deformities were shown beyond 90 months p.i. In this animal, antineural antibodies were detectable about 30 months before clinical nerve damage. Of particular interest, antibody to nerve antigens peaked about 5 months prior to the recurrence of clinical leprosy after a spontaneous remission of the initial disease at 9 months p.i.
Mangabey monkeys D173 and D174 were inoculated with 4.8 x 109 M. leprae. In mangabey D173, early lesions of leprosy appeared 5 months p.i. but regressed by 14 months. At 86 months p.i., the first neurologic damage and bone absorption were noted, followed by progression of lepromatous (LL) leprosy. The animal required chemotherapy at 105 months p.i. This animal had significant IgM antibodies to AGM1 before inoculation for unknown reasons, and had small fluctuations with low levels of antibodies (Fig. 2A). Meanwhile, there was a slight increase in IgG and IgM antibodies to ceramide at 3 months, but no major antibody response was shown until 63 months p.i. at which time there was a strong IgG antibody response to ceramide.
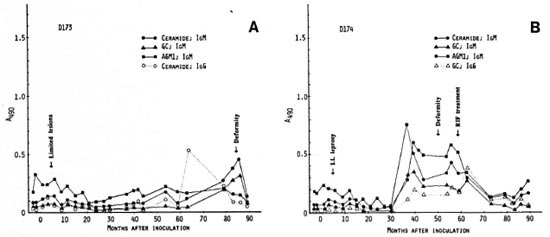
Fig. 2. Antibody responses to nerve antigens in mangabeys D173 (A) and D174 (B) before and at intervalsafter inoculation with 4.8 x 109 M. leprae .
This was followed by a steady increase in IgM antibodies to ceramide and GC antigens which peaked at 85 months p.i., just 1 month before the first sign of neurologic damage. However, the IgM antibodies declined sharply within 4 months after onset of clinical nerve damage in the absence of chemotherapy. In this animal, therefore, a major antineural antibody peak was noted about 23 months prior to clinical nerve damage.
Mangabey D174 developed Iepromatous (LL) leprosy by 10 months p.i. but regressed at 14 months. However, clinical signs of leprosy recurred in this animal at 35 months p.i. and persisted, requiring chemotherapy at 59 months. Neurologic deformities became obvious at 50 months and worsened after the initiation of chemotherapy. In this monkey, moderate levels of IgM antibodies to all three antigens appeared at 36 months p.i., coinciding with clinical reactivation, and were maintained until 63 months (Fig. 2B). Thus, antineural antibodies were detectable about 14 months earlier than neurological deformities, and the antibody levels decreased shortly after starting chemotherapy.
Mangabey D175, inoculated with 4.8 X 108 M. leprae, developed no clinical leprosy to date, 9 years p.i. There also has been no significant antibody response to the nerve antigens under investigation except for a small peak of IgM antibodies to ceramide at 59 months p.i. (Fig. 3A).
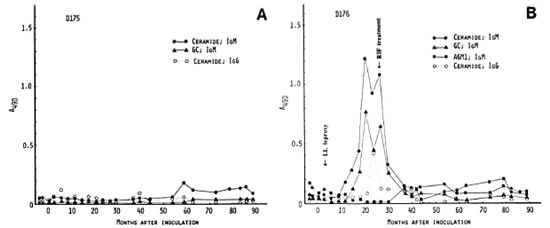
Fig. 3. Antibody responses to nerve antigens in inangabeys D175 (A) and D176 (B) before and at intervalsafter inoculation with 4.8 x 108M. leprae.
In mangabey D176, also inoculated with 4.8 X 108 M. leprae, Iepromatous (LL) leprosy was noted at 4 months p.i., and the disease was extensive by 15 months. At 27 months p.i., rifampin treatment was initiated, resulting in complete remission at 35 months. There were strong IgM responses to ceramide and GC antigens at 15 months which lasted until 27 months p.i. (Fig. 3B). There were also small IgM antibody peaks to AGM1 during the study period. Despite these strong antineural antibody responses, however, no neurological signs have been observed.
Mangabeys D177 and D178 received 4.8 X 107 M. leprae. In mangabey D177, there was a slow development of clinical leprosy (LLs) by 26 months p.i. At 42 months, treatment with rifampin was started. Nerve enlargement was first detectable at 47 months p.i. and persisted after successful chemotherapy. There were minor peaks of antibodies to ceramide which were followed by a strong IgG-antibody peak at 40 months p.i. (Fig. 4A). The antibodies to ceramide and GC declined after starting chemotherapy. Thus, antineural antibody responses appeared 7 months prior to palpable nerve enlargement.
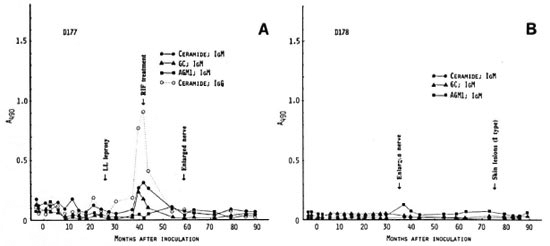
Fig. 4. Antibody responses to nerve antigens in mangabeys D177 (A) and D178 (B) before and at intervalsafter inoculation with 4.8 x 107 M. leprae .
In mangabey Dl 78, an enlarged nerve was the first sign of leprosy at 35 months p.i. Histopathologically, there were indeterminate lesions of leprosy in the skin which later regressed spontaneously. However, neurological damage continued beyond 105 months. No chemotherapy was given. Despite the presence of nerve enlargement, there were no major antibody responses to the three neural antigens except for a small peak of IgM antibody to AGM1 at 37 months p.i., 2 months after the nerve enlargement was noticed (Fig. 4B).
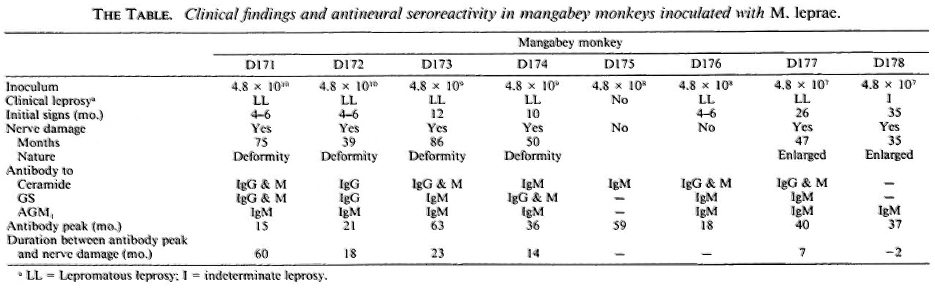
The observations obtained in the eight animals are summarized in The Table. Of 8 mangabey monkeys inoculated with M. leprae, 7 animals had clinical leprosy; 6 of them had clinical evidence of damage to nerves-neurologic deformities and nerve enlargement. The median time for the initial signs of leprosy was 10 months (range 4 to 35 months). In contrast, nerve damage was noted rather late, about 35 to 86 months p.i. (median 54 months). The major immunoglobulin class to ceramide, GC and AGM1 antigens was IgM, and the antibody responses to the nerve antigens were detectable from 15 to 63 months p.i. (median 37 months). Antineural antibodies were detectable at a mean of about 15 months (range - 2 to 60 months). Thus, antineural antibodies appeared long before clinical evidence of nerve damage.
DISCUSSION
In this study, we present clear evidence that antineural antibodies are produced during the course of M. leprae infection. Consequently, the antineural antibodies appear to be indicative of future damage to nerves with neuropathic deformities. After inoculation of mangabey monkeys with M. leprae, humoral antibody responses to M. leprae antigens, such as phenolic glycolipid (PGL)-I and lipoarabinomannan (LAM), appeared relatively early (3 to 6 months after infection 6,7). In contrast, the median time for the first major peaks of antineural antibodies was 37 months p.i. (range 15 to 63 months). This was a rather late response considering that the initial signs of clinical leprosy appeared 5 to 10 months p.i. in the majority of the monkeys. This may, in turn, indicate that destruction of neural tissue proceeds slowly and, perhaps, is related to the slow growth rate of M. leprae. Once the nerve tissues are damaged by mechanisms which remain to be determined, the host's immune system appears to initiate antibody responses to the nerve antigens.
Of particular interest is that high levels of antibodies to nerve antigens were strongly associated with the onset and progress of clinical leprosy. For example, strong antineural antibody responses were found during rapid progression of the disease or development of extensive disease in mangabeys D171, D174, D176, and D177. In addition, an increase in antineural antibodies preceded by several months the recurrence of clinical leprosy or neurologic signs, as shown in mangabeys D172, D173 and D174. Therefore, one can assume that during the early stage of M. leprae infection, the bacterial load is not large and, despite clinical signs of leprosy, nerve involvement may be very limited and, thus, may not be detectable during regular physical examination. thus, by detecting antineural antibodies, it may be feasible to predict future clinical eruption of leprosy and/or nerve damage in persons infected with M. leprae.
there also seem to be several fluctuations of antineural antibody levels during the course of M. leprae infection in the absence of chemotherapy, as shown in mangabeys D171, D172, D173, AND D177. this may imply that nerve tissues are destroyed intermittently, thus boosting antibody responses to the antigens. thi s suggests that there ma y be a wide variation in antineural antibody reactivity in sera from leprosy patients at the time of diagnosis, depending on the natural history of M. leprae infection s: in each patient.
In leprosy-endemic areas, many new leprosy patients are diagnosed with extensive disease long after initial signs have appeared. since nerve damage is one of the cardinal signs of leprosy, almost all new leprosy patients have more or less nerve involvements, such as anesthesia, nerve enlargements and/or neurologic deformities, at the time of diagnosis (3,18). Therefore, it is not surprising that 40%-70 % of new leprosy patients had elevated antibodies to ceramide, GC , OR AGM1 antigen. In addition, antineural antibodies are strongly associated with the extent of nerve involvement in leprosy patients at the time of diagnosis (24; Cho, et al, unpublished data).
As shown in this study, antineural antibodies were studied in leprosy patients in an attempt to elucidate the pathogenic mechanism(s) of nerve damage. This hyc pothesis was based on the concept that the antibodies may be directly involved in nerve tissue destruction, such as in Guillain-Barre syndrome (20 21), motor neuron diseases (16,17), systemic lupus erythematosus with neurological disorders (8,19) , and multiple sclerosis (1). Antibodies to GC, AGM1, or other gangliosides have been reported to play a role in demyelination of the nerves in these diseases. However, although abnormal my lin changes were observed in nerve fibers (9), demyelination is not a prominent feature in leprosy. Rather, delayed-type hypersensitivity to m. leprae antigens in nerve tissues may provoke granuloma formation and destroy the nerves (10,15). Once the nerves arc damaged, humoral antibody responses to nerve tissue antigens would undoubtedly follow, resulting in high antineural antibody levels.
During hypersensitivity as seen in lepra reactions (e.g., erythema nodusum leprosum or reversal or upgrading reactions), immediate nerve pain or damage will be noted, but in a majority of leprosy patients, a slow progress of nerve destruction will occur (12) and eventually result in neurologic deformity, as seen in experimentally infected mangabey monkeys in this study. Therefore, despite hypersensitivity perhaps being the initial trigger of nerve damage along with M. leprae growth in the tissues, antineural antibodies still may be associated with the autoimmune-mediated destruction of nerve trunks leading to neurological deformities or nerve enlargement (22). Further study is warranted to explain the exact role of antineural antibodies in the pathogenesis of leprosy. Meantime, the detection of antibodies to nerve tissue antigens may be used for the assessment of leprosy patients by providing information on the degree of nerve tissue damage. Antineural antibody reactivity could be used as a parameter for the diagnosis of leprosy if combined with tools detecting M. leprae infection using M. lep rac-specific antigens such as PGL-I (4).
In this study, we inoculated a relatively large number of M. leprae compared to the dose presumed to be involved in the natural transmission of leprosy between animals or between leprosy patients and their contacts. Therefore, we do not know how to extrapolate precisely the clinical course of leprosy from these mangabey monkeys to human leprosy. However, as far as antibody responses to nerve antigens are concerned, this study produced unequivocal evidence that antineural antibodies usually preceded neurologic deformities or other neuropathologic events. It remains to be determined if these antibodies to nerve antigens are just bystander responses or if they play a role in the pathogenesis of nerve damage.
Acknowledgment. This work was supported in part by grants from the UNDP/World Bank/WHO Spécial Programme for Research and Training in Tropical Diseases and from the National Institutes of Health grants #AI 19302 from the National Institutes of Allergy and Infectious Diseases and #RR00164 from the Animal Resources Program, National Center for Research Resources. The authors thank E. O. Shin and M. K. Lee for technical assistance.
REFERENCES
1. ARNON, R., CRISP, E., KELLEY, R., ELLISON, G. W., MYERS, L. W. and TOURTELLOTTE, W. W. Antigangliosidc antibodies in multiple sclerosis. J. Neurol. Sci. 46(1980)179-186.
2. BENJAMINS, J. A., CALLAHAN, R. E., RUNFT, D., GERRAS, G. and LEFFORD, M. J. Anti-neural antibodies in leprosy sera: further characterization of the antigens. J. Neuroimmunol. 21(1989)125-135.
3. CHAROSKY, C. B., GATTI, J. C. and CARDAMA, J. E. Neuropathies in Hansen's disease. (Editorial) Int. J. Lepr. 51(1983)576-586.
4. CHO, S.-N., YANAGIHARA, D. L., HUNTER, S. W., GELBER, R. H. and BRENNAN, P. J. Serological specificity of phenolic glycolipid I from Mycobacterium leprae and use in serodiagnosis of leprosy. Infect. Immun. 41(1983)1077-1083.
5. GHASWALA, P. S., MISTRY, N. F. and ANTIA, N. H. Serum antibodies of normals and leprosy patients show equal binding to peripheral nerve. Int. J. Lepr. 57(1989)690-692.
6. GORMUS, B. J., OHASHI, D. K., OHKAWA, S., WALSH, G. P., MEYERS, W. M., BRENNAN, P. J. and TRYGG, C. Serologic responses to Mycobacterium leprae specific phenolic glycolipid-I antigen in sooty mangabey monkeys with experimental leprosy. Int. J. Lepr. 56(1988)537-545.
7. GORMUS, B. J., Xu, K., MEYERS, W. M., WALSH, G. P., LEVIS, W. R. and MEEKER, H. C. Antibodies to lipoarabinomannan antigen in sooty mangabey monkeys experimentally inoculated with Mycobacterium leprae. Int. J. Lepr. 58(1990)65-72.
8. HIRANO, T., HASHIMOTO, H., SHIOKAWA, Y., IWA-MORI, M., NAGAI, Y., KASAI, M., OCHIAI, Y. and OKUMURA, K. Antiglycolipid autoantibody detected in the sera from systemic lupus erythematosus patients. J. Clin. Invest. 66(1980)1437-1440.
9. JACOBS, J. M., SHETTY, W. P. and ANTIA, N. H. Myelin changes in leprous neuropathy. Acta Ncuropathol. 74(1987)75-80 .
10. JOB, C. K. Mechanism of nerve destruction in tuberculoid-borderline leprosy; an electron-microscopic study. J. Neurol. Sci. 20(1973)25-38 .
11. JOB, C. K. Nerve damage in leprosy. Int. J. Lepr. 57(1989)532-539.
12. JOB, C. K., VICTOR, D. B. I. and CHACKO, C. J. G. Progressive nerve lesion in a disease-arrested leprosy patient; an electron micorscopic study. Int. J. Lepr. 45(1977)255-260.
13. KIRCHHEIMER, W. F. and STORRS, E. E. Attempts to establish the armadillo (Dasypus novemcinctus Linn.) as a model for the study of leprosy. 1. Report of lepromatoid leprosy in an experimentally infected armadillo. Int. J. Lepr. 39(1971)693-702.
14. MSHANA, R. N., HARBOE, M., STONER, G. L., HUGHES, R. A. C., KADLUBOWSKI, M. and BELEHU, A. Immune responses to bovine neural antigens in leprosy patients. I. Absence of antibodies to an isolated myelin protein. Int. J. Lepr. 51(1983)33-40.
15. MSHANA, R. N., HUMBER, D. P., HARBOE, M. and BELEHU, A. Nerve damage following intraneural injection of Mycobacterium leprae into rabbits pre-sensitized to mycobacteria. Clin. Exp. Immunol. 52(1983)441-448.
16. NOBILE-ORAZIO, E., CARPO, M., LEGNAME, G., MEUCCI, N., SONNINO, S. and SCARLATO, G. Anti-GM1 IgM antibodies in motor neuron disease and neuropathy. Neurology 40(1990)1747-1750.
17. PESTRONK, A., CHAUDHRY, V., FELDMAN, E. L., GRIFFIN, J. W., CORNBLATH, D. R., DENYS, E. H., GLASBERG, M., KUNCL, R. W., OLNEY, R. K. and YEE, W. C. Lower motor neuron syndromes defined by patterns of weakness, nerve conduction abnormalities, and high titers of antiglycolipid antibodies. Ann. Neurol. 27(1990)316-326.
18. RIDLEY, D. S. and JOPLING, W. H. Classificationof leprosy according to immunity; a five-groupsystem. Int. J. Lepr. 34(1966)255-273.
19. ROBBINS, M. L., KORNGUTII, S. E., BELL, C. L.,KALINKE, T., ENGLAND, D., TURSKI, P. andGRAZIANO, F. M. Antineurofilament antibodyevaluation in neuropsychiatric systemic lupus er-ythematosus. Arthritis Rheum. 31(1988)623-631.
20. RYBERG, 13., HINDFELT, B., NILSSON, B. and OLSSON,J. E. Antincural antibodies in Guillain-Barre syndrome and lymphocytic meningoradiculitis (Bannwarth's syndrome). Arch. Neurol. 41(1984)1277-1281.
21. SAIDA, T., SAIDA, K., LISAK, R. P., BROWN, M. J.,SILBERBERG, D. H. and ASBURY, A. K. In vitro demyelinating activity of sera from patients with Guillain-Barrê syndrome. Ann. Neurol. 11(1982)69-75.
22. SHETTY, V. P., MISTRY, N. F. and ANTIA, N. H.Serum demyelinating factors and adjuvant-like activity of Mycobacterium leprae: possible causes ofearly nerve damage in leprosy. Lepr. Rev. 56(1985)221-227.
23. THOMAS, B. M. and MUKHERJEE, R. Antineural antibodies in sera of leprosy patients. Clin. Immunol. Immunopathol. 57(1990)420-429.
24. VEMURI, N. and MUKHERJEE, R. Immunoreactivity of nerve lipid antigens in leprosy. J. Clin. Lab. Anal. 5(1991)157-161.
25. VOLLER, A., BinwELL, D. E. and BARTLETT, A. The Enzyme-linked Immunosorbent Assay (ELISA) . Alexandria, Virginia, U.S.A.: Dynatech Laboratories, Inc., 1979.
26. WOLF, R. H., GORMUS, B. J., MARTIN, L. N., BAS-KIN, G. B., WALSH, G. P., MEYERS, W. M. andBINFORD, C. H. Experimental leprosy in threespecies of monkeys. Science 227(1985)529-531.
27. WRIGHT, D. J. M., HIRST, R. A. and WATERS, M.F. R. Neural autoantibodies in leprosy. Lepr. Rev. 46(1975)157-169.
1. D.V.M., Ph.D.; Department of Microbiology, Yonsci University College of Medicine, C.P.O. Box 8044, Seoul 120-752, Republic of Korea.
2. M. D., Ph.D., Department of Microbiology, Yonsci University College of Medicine, C.P.O. Box 8044, Seoul 120-752, Republic of Korea.
3. Ph.D.; Tulane Regional Primate Research Center, Covington, Louisiana 70433, U.S.A.
4. M.D., Department of Microbiology; Tulane Regional Primate Research Center, Covington, Louisiana 70433, U.S.A.
5. D. V.M., Department of Veterinary Sciences, Tulane Regional Primate Research Center, Covington, Louisiana 70433, U.S.A.
6. Ph.D., Leonard Wood Memorial Center for Leprosy Research, Cebu, The Philippines.
7. M.D., Ph.D., Armed Forces Institute of Pathology, Washington, D.C. 20306-6000, U.S.A.
Received for publication on 30 November 1992.
Accepted for publication in revised form on 24 March 1993.