- Volume 61 , Number 3
- Page: 406–14
Activity of phenazine analogs against Mycobacterium leprae infections in mice
ABSTRACT
Twenty-five compounds structurally related to clofazimine were tested for their ability to inhibit the growth of Mycobacterium leprae using the kinetic method of drug evaluation in the mouse foot pad model of leprosy. Seven of the phenazine derivatives displayed anti-M. leprae activity comparable to that of clofazimine when administered at a concentration of 0.01% (w/w) in the diet. Three of the compounds, B746, B4087, and B4101, were active when administered at 0.001% in the diet. At a dietary concentration of 0.0001%, B4087 and B4101 were slightly more active than clofazimine, while B746 was less active. In the kinetic method of drug evaluation, greater anti-M. leprae activity of phenazine derivatives was generally associated with greater pigmentation of abdominal fat. Of the compounds which did not cause pigmentation when fed at a concentration of 0.01% in the diet, B4090 was the most active. This compound also inhibits the growth of a clofazimine-resistant M. smegmatis strain.RÉSUMÉ
Vingt-cinq composes structurellement apparentés á la clofazimine ont été testes, par la méthode cinétique de revaluation des medicaments dans le modele du coussinet plantaire de la souris, quant á leur capacité de limiter la croissance de Mycobacterium leprae. Sept des derives phenazine ont montré une activité comparable à celle de la clofazimine quand administrés à une concentration de 0.01% (p/p) dans le régime alimentaire. Trois des composés, B746, B4087 et B4101, étaient actifs à la concentration de 0.001% dans le régime alimentaire. A une concentration de 0.0001%, B4087 et B4101 étaient légèrement plus actifs que la clofazimine, tandis que B746 l'était moins. Dans la méthode cinétique d'évaluation des médicaments, une activité anti-M. leprae plus grande des dérivés phenazine était généralement associée à une pigmentation plus prononcée de la graisse abdominale. Parmi les composés qui ne causaient pas de pigmentation quand administrés à une concentration de 0.01% dans le régime alimentaire. B4090 était le plus actif. Ce composé inhibe également la croissance d'une souche de M. smegmatis résistante à la clofazimine.RESUMEN
Utilizando el método cinético de evaluación de drogas en el modelo de la almohadilla plantar del ratón, se estudiaron 25 compuestos estructuralmente relacionados a la clofazimina para probar su capacidad de inhibir el crecimiento de Mycobacterium leprae. Siete de los derivados de la fenazina, administrados en la dieta a una concentración del 0.01%, mostraron actividad anti-M. leprae comparable a la de la clofazimina. Tres de los compuestos, B746, B4087, y B4101. fueron activos cuando se administraron a la concentración de 0.001%. A la concentración dictaría de 0.0001%. mientras que B4087 y B4101 fueron ligeramente más activos que la clofazimina, el B746 fue menos activo. También se notó que mientras mayor fue el efecto anti-M. leprae de los derivados de la fenazina, mayor fue su asociación con la pigmentación de la grasa abdominal. Entre los compuestos que no causaron pigmentación cuando se administraron a la concentración dictaría de 0.01%, el B4090 fue el más activo. Este compuesto también inhibió el crecimiento de una cepa de M. smegmatis resistente a clofazimina.Hansen's disease (leprosy) is a chronic, infectious disease primarily affecting the skin and peripheral nerves which can result in debilitating deformities if untreated. The World Health Organization (WHO) estimates that Hansen's disease afflicts 5.5 million people worldwide, mainly in developing countries (6). In 1981, WHO first recommended the use of multidrug regimens, which include dapsone, rifampin, and clofazimine, to prevent the emergence of drug-resistant strains of Mycobacterium leprae and to effectively shorten the period of chemotherapy (14). Both primary and acquired resistance to dapsone and rifampin have been reported (13,14 ) . No strains of M. leprae resistant to clofazimine have been reported even though it has been used as monotherapy in many patients (13).
Clofazimine (also designated B663) is a 3-(p-chloroanilino)- lO-(p-chlorophenyl)2,10-dihydro-2-(isopropylimino) phenazine which was first synthesized in 1954 by Barry and colleagues (1). They found it to be one of the most active of a series of phenazine compounds against M. tuberculosis in animal studies (2), and subsequent studies demonstrated its anti-M leprae activity in mice (l2) and humans (3). Clofazimine is a highly fat soluble compound which is deposited in the tissues and is slowly eliminated from the body. It has the common, undesirable side effects of a dose-related skin pigmentation and a dose-limiting gastrointestinal toxicity (15).
O'Sullivan and colleagues have identified clofazimine analogs that are less soluble in fat and more readily excreted than clofazimine, do not crystalize within macrophages, and are active against a clofazimine-resistant strain of M. smegmatis (7; JOS, unpublished results). In general, these phenazine derivatives differ from clofazimine by having a basic nitrogen group attached via a bridge of at least three carbon atoms to the imino-nitrogen (R3) of the phenazine nucleus (Table 1). To determine which of the phenazine derivatives were active against M.leprae, Franzblau, et al. (4,5) analyzed them in vitro using a radiometric metabolic assay. Their initial study included 12 phenazines that were active against the clofazimine-resistant M. smegmatis strain and that differed from clofazimine in containing a basic nitrogen attached to the imino-nitrogen (R3) and, in some cases, in chlorination of the phenazine nucleus (Rl) or at the para positions of the aniline and phenyl rings (R2) (Table 1). They found that the anti-M. leprae activity of these compounds in the in vitro assay generally increased with the degree of chlorination at the Rl and R2 positions and that phenazines with a 2,2,6,6tetramethylpiperidine (TMP) substitution at the imino-nitrogen were the most active of the compounds studied and, indeed, were more active than clofazimine (4). In a subsequent study with an additional five derivatives of the TMP analog, Franzblau, et al. (5) found that the anti-M. leprae activity increased in the TMP derivatives with substitutions of the hydrogens on the phenyl and aniline groups (R2) by fluorines, ethoxy groups, methyl groups, chlorines, and bromines in that order. The TMP analog displaying the greatest anti-M. leprae activity was B4076.

In the studies reported here, clofazimine and 25 phenazine derivatives were surveyed for their activity against experimental M. leprae infections in mice. The studies included most of the analogs assayed in the in vitro system. Twenty-two of the analogs contain a basic nitrogen group on the imino-nitrogen and are active against the clofazimine-resistant M. smegmatis strain. These in vivo studies are a necessary complement to the in vitro studies because M. leprae is an intracellular pathogen and anti-M. leprae activity in vivo will be influenced by pharmacokinetic factors.
MATERIALS AND METHODS
Antimicrobial agents. The basic phenazine structure is shown in Figure 1. The chemical groups at the R1, R2, and R3 positions of the phenazine for each of the 25 compounds surveyed and clofazimine are shown in Table 1. These compounds were synthesized at Trinity College, Dublin, aspreviously described (1).
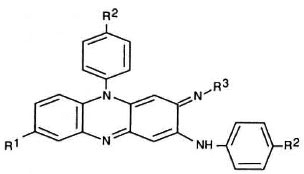
Fig. 1. Basic phenazinc structure.
Strains. Five strains of M. leprae were used in these studies. Strains B2602, B2652, B2409, and B2439 were obtained from the U.S. Public Health Service Hospital, Carville, Louisiana, U.S.A. Each strain is from an untreated lepromatous patient, except strain B2602 which is from a patient who had received 1 year of dapsone therapy 2 years before admission. Strains B2602 and B2439 are from patients from Puerto Rico; strain B2652 is from a patient from Okinawa; B2409 is from a patient from the United States; and strain B3211 is from a patient of Philippine origin. All strains have been maintained in regular mouse passage at the Centers for Disease Control, Atlanta, Georgia, U.S.A. None of the strains have any known drug resistances.
Drug evaluation in vivo. The kinetic method of drug evaluation was used to determine the activity of the compounds against M. leprae in vivo as previously described (11). Briefly, approximately 5000 acid-fast M. leprae bacilli were injected in the right hindfoot pad of 3-week-oldfemale CFW mice. The drugs were mixed with ground commercial mouse chow (Rodent Lab Chow 5001; Purina Mills, Inc., Richmond, Indiana, U.S.A.) in a twin-shell blender (Patterson Kelly Co., East Stroudsburg, Pennsylvania, U.S.A.) at concentrations of 0.01% and 0.001% (w/w). A concentration of 0.001 % in the diet corresponds to a dosage of about 1.0 mg/kg/day. Mice were fed the drug-containing diets from day 70 after inoculation through day 126. Three of the drugs were also tested at a concentration of 0.0001% (w/w). Control groups were fed diets containing either no antimicrobial agents or clofazimine at the same concentrations as above.
Growth of M. leprae in the foot pads of control mice fed a drug-free diet was measured at 28-day intervals beginning on day 70 by enumerating bacilli in a pool of foot pad tissue collected from four mice. When the growth in this control group reached approximately 106 bacilli per mouse or plateau (two successive counts at > 5 × 105 bacilli per mouse), the foot pads of five mice in each group were processed individually and the number of bacilli per foot pad were enumerated. Subsequently, counts were repeated at intervals of about 4 months until the number of bacilli per foot pad for the treated group was equal to or greater than that for the untreated control group, or until no more mice were available. The numbers of bacilli per mouse in the various groups were compared statistically using the Wilcoxon rank sum test. Using this method the probability (p) that the difference in the observed results for two groups is due solely to random fluctuations in the data was calculated (11). Differences with a p value of < 0.05 are considered statistically significant. At each harvest, the color of the abdominal fat was also noted.
The magnitude of inhibition of M. leprae in the treated mice was determined by calculating the number of days between the growth curve in the untreated control group and a parallel growth curve drawn for the treatment groups. Delays of growth could be calculated only for compounds which showed a statistically significant inhibition of growth (p < 0.05) in one or more of the timepoints. A delay of "> x" days indicated that no statistically significant growth was observed in the group as of the last timepoint; "x" was determined by assigning a value of 8 × 104 bacilli per foot pad (the minimum number detectable in our system) to the group and calculating growth delay as described above.
RESULTS
As found in previous studies using the kinetic method of drug evaluation (8), the inhibition of M. leprae growth by clofazimine was significant (p < 0.05) when it was mixed in the diet at concentrations of 0.01 %, 0.001%, and 0.0001% (w/w). Significant inhibition of growth was found with 17 of the 25 compounds tested at a concentration of 0.01% in the diet (Table 2, Fig. 2). Only six of these compounds significantly inhibited the growth of M. leprae when tested at a concentration of 0.001 %. Three compounds (B746, B4087, B4101) were also inhibitory when tested at a dietary concentration of 0.0001%. The remaining eight compounds (B825, B3752, B3768, B3849, B3935, B3952, B4017, B4018) were not inhibitory when tested at concentrations of 0.01% and 0.001% in the diet.
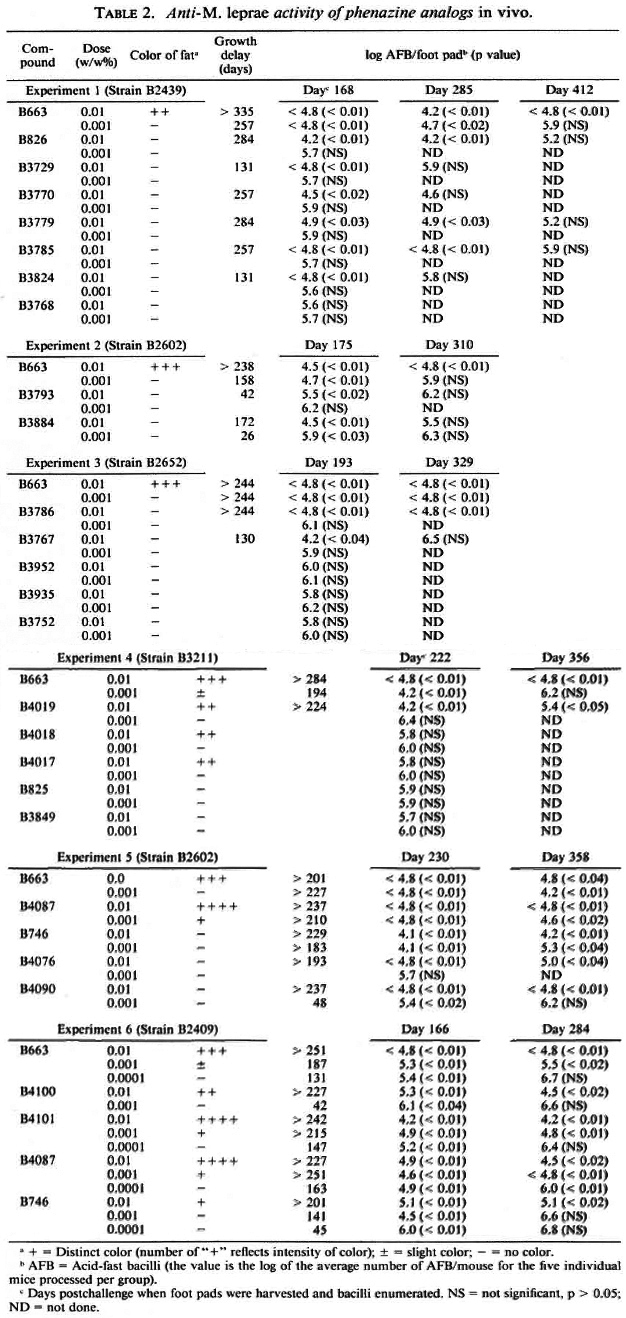
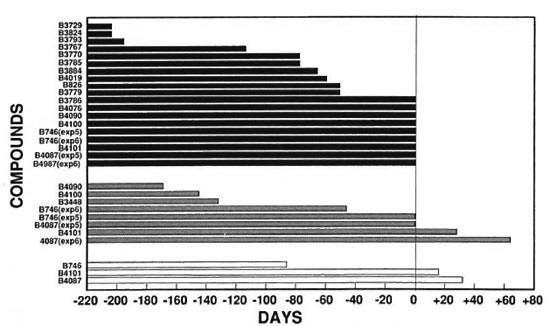
Fig. 2. Growth delays of compounds tested relative to clofazimine (13663). The number of days the growth delay of compounds tested differed from clofazimine (B663) was calculated by subtracting the number of days of growth delay for each compound from the number of days of growth delay for the same concentration of clofazimine (B663) in the respective experiment: ■ = relative to 0.01% cloratimine (13663)  = relative to 0.001% clofazimine (B663); □ = relative to 0.0001% clofazimine (13663).
= relative to 0.001% clofazimine (B663); □ = relative to 0.0001% clofazimine (13663).
In the six separate experiments, delays of M.leprae growth in the foot pads of mice that received 0.01% clofazimine in the diet varied from > 201 days to > 335 days (Table 2). Because of this variability, comparisons of activities were made only for compounds included in the same experiment. Overall, the delays of growth in the foot pads of mice that received the phenazine analogs at a dietary concentration of 0.01% varied from substantially less than (B3729, B3767, B3793, B3824) to slightly less than (B3884, B826, B3770, B3779, B3785, B4019) to comparable to (B746, B3786, B4087, B4076, B4090, B4100, B4101) that observed in the groups receiving 0.01% clofazimine (Table 2, Fig. 2).
The growth delays in the foot pads of mice receiving 0.001% clofazimine in the diet varied from 158 days to 257 days (Table 2). The growth delays in the foot pads of mice that received 0.001% of the phenazine analogs in the diet varied from none (B826, B3729, B3767, B3770, B3779, B3785, B3786, B3793, B3824, B4019, B4076) to substantially less than (B3884, B4090, B4100) to somewhat less than (B746) to comparable to or slightly greater than (B4087, B4101) that observed in the groups receiving 0.001% clofazimine (Table 2, Fig. 2).
The delay of growth in the foot pads of mice that received 0.0001% clofazimine in the diet was 131 days. The delay in the foot pads of mice that received this concentration of B746 in the diet was 45 days, of B4101 147 days, and of B4087 163 days (Table 2). Also, the numbers of bacilli per foot pad in the last two treatment groups were significantly less than that in the group fed 0.0001% clofazimine.
Distinctly pigmented abdominal fat was observed in mice that received B4017, B4018, B4019, B4087, B4100, B4101, or clofazimine at a concentration of 0.01% in the diet (Table 2). B4017, B4018, and B4019 also strongly pigmented intestinal tissues. Mice that received 0.01% B746 in the diet had moderately pigmented fat in one experiment and no pigmentation in a second experiment. Pigmentation in mice fed diets containing 0.01 % B4087 or B4101 was more intense than in mice fed diets containing 0.01% clofazimine, which was more intense than in mice fed diets containing 0.01% B4017, B4018, B4019, or B4100. Pigmentation in mice fed diets containing 0.001% B4087 or B4101 was also greater than that in mice fed diets containing 0.001% clofazimine. When a compound produced pigmentation at both the 0.01% and 0.001% concentrations, the degree of pigmentation was dose related. No pigmentation was observed in any of the animals that received drugs at 0.0001%.
DISCUSSION
Seventeen of the clofazimine derivatives showed some anti-M. leprae activity in vivo, and this activity generally paralleled the activity seen in vitro (4, 5). For example, within the series of compounds having an active basic group attached to the imino-nitrogen through a propanyl spacer group and the same groups at the Rl and R2 positions, compounds having a diethylamine, pyrrolidine ring, or piperidine ring at R3 were about equally active, and they were more active than those with a diethanolamine at R3 both in vivo and in vitro. Also, within series of derivatives clustered by shared substitutions at R3, the most active compound in vitro (5) was also the most active in vivo.
One significant difference between the in vivo and in vitro results concerns the effect of chlorination at the Rl and R2 positions. Within a series of compounds with the same R3 group, the anti-M. leprae activity in vitro increased with the degree of chlorination at either the Rl or R2 positions. In contrast, in the in vivo studies, activity increased only with the degree of chlorination at the para positions of the aniline and phenyl rings (R2). Furthermore, three of the four compounds with a chlorine at the Rl position of the phenazine nucleus and hydrogens at the R2 positions were inactive in vivo, although highly active in vitro. The one which was active both in vivo and in vitro (B4019) displayed much greater activity in vitro than the other three compounds and was the only one of the four to pigment abdominal fat.
Of the compounds effective against the clofazimine-resistant M. smegmatis strain, the TMP derivatives displayed the greatest anti-M. leprae activity both in vivo and in vitro, and the activity in vivo generally paralleled the activity in vitro. However, in marked contrast to the results in vitro, the TMP derivatives were much less effective than clofazimine against M. leprae in vivo. Similarly, the three compounds with an R3 substitution chemically similar to that of 13663 were more active than the TMP derivatives in vivo, but less active in vitro (JOS, unpublished results).
The differences between the results in vivo and in vitro may reflect the influence of pharmacokinetics and the nuances of the kinetic method of drug evaluation. In the kinetic method, drugs that delay bacterial growth for a period of time longer than drug administration act by one of three mechanisms (10): bactericidal activity, bacteripausal activity (i.e., persisting bacteristasis after the elimination of the drug), or a repository effect (i.e., persistence of the drug in the tissues at inhibiting concentrations after administration is stopped). Clofazimine produces very long growth delays in mice (8-10). It is likely that these long delays are due to the persistence of clofazimine in the mice because clofazimine accumulates in fat and tissues, crystalizes in macrophages, and is excreted slowly (8,9).
The most active compounds in vivo were the ones producing the greatest pigmentation, and there was a positive correlation between the degree of pigmentation and the delay of bacterial growth. (The correlation, however, was not absolute in that two compounds, B4017 and B4018, pigmented abdominal fat but did not inhibit the growth of M. leprae) The two compounds, B4087 and B4101, that produced delays greater than that of clofazimine also produced more intense and more consistent pigmentation of abdominal fat than clofazimine. The greater anti-M. leprae activity of the more highly pigmenting compounds may reflect greater accumulation and persistence of these compounds in fat and tissue rather than, or in addition to, greater intrinsic activity against M. leprae. Intrinsic anti-M. leprae activity as measured in the in vitro assay (4-5) is also important because among the group of compounds that did not stain abdominal fat, compounds that were more active in vitro were also generally more active in vivo. Overall, the results indicate that while pigmentation is not a prerequisite for a clofazimine-related compound to be effective against M. leprae infection in mice, the more active phenazine compounds in the kinetic method of drug evaluation did cause more pigmentation.
In contrast to clofazimine, most of the TMP derivatives are less soluble in fat, do not form crystals in macrophages, and are excreted more rapidly (JOS, unpublished results). They also caused less pigmentation and were less active in the kinetic method of drug evaluation. Nonetheless, all five TMP derivatives tested strongly inhibited the growth of M. leprae when fed at 0.01% in the diet and two, B4090 and B4100, displayed modest anti-M. leprae activity at a dietary concentration of0.001%. B4090 and B4100 are the most active of the TMP derivatives in the in vitro assay (JOS, personal communication). B4090 did not pigment abdominal fat when fed at 0.01% or 0.001% in the diet, while B4100 did pigment abdominal fat when fed at 0.01%. Interestingly, the 3,4 dichlorine substitutions were made at the R2 positions in B4100 to increase fat solubility.
Three of the TMP derivatives (B3786, B4019, and B4090) have been fed continuously to mice at 0.001% in the diet, and all three inhibited the growth of M. leprae (JOS, unpublished results). This concentration of drug in the diet corresponds to a dose of approximately 1 mg/kg/day. This is roughly equivalent to the dosage used to treat Hansen's disease in humans. Since B3786 and B4090 are nonpigmenting and active against the clofazimine-resistant strain of M. smegmatis, they are candidates for nonpigmenting clofazimine analogs which might also be active against a clofazimine-resistant M. leprae strain should one arise.
REFERENCES
1. BARRY, V. C, BELTON, J. G., CONALTY, M. L., DENNENY, J. M.. EDWARD, D. W., O'SULLIVAN, J. F., TWOMEY, D. and W IDNER, F. A new scries of phenazine (rimino-compounds) with a high antituberculosis activity. Nature 179(1957)1013-1015.
2. BARRY, V. C. and CONALTY, M. L. Antituberculosis activity in the phenazine scries. II. N3-substituted anilinoaposafranines (rimino-compounds) and some derivatives. Am. Rev. Tuberc. Pulm. Dis. 78(1958)62-73.
3. BROWNE. S. G. and HOGERZEIL, L. M. B663 in the treatment of leprosy; a preliminary report of a pilot trial. Lepr. Rev. 33(1962)6-10.
4. FRANZULAU, S. G. and O'SULLIVAN, J. F. Structure-activity relationships of selected phenazines against Mycobacterium leprae in vitro. Antimicrob. Agents Chemother. 32(1988)1583-1585.
5. FRANZULAU, S. G., WHITE, K. E. and O'SULLIVAN, J. F. Structure-activity relationships of tetramethylpiperidine-substituted phenazines against Mycobacterium leprae in vitro. Antimicrob. Agents Chemother. 33(1989)2004-2005.
6. N OORDEEN, S. K., B RAVO, L. L. and S UDARESAN, T. K. Estimated number of leprosy cases in the world. Bull. WH O 70(1992)7-10.
7. O'S ULLIVAN. J. F., CONALTY, M. L. and MORRISON, N. E. Clofazimine analogues active against a clofazimine-resistant organism. J. Med. Chem. 31(1988)567-572.
8. SHEPARD. C. C. Minimal effective dosages of clofazimine (B663) and of ethionamide against Mycobacterium leprae. Proc. Soc. Exp. Biol. Med. 132(1969)120-124.
9. SHEPARD, C. C. A survey of the drugs with activity against M. leprae. Int. J. Lepr. 39(1971)340-348.
10. SHEPARD, C.C. Combinations involvingdapsone, rifampin, clofazimine, and ethionamide in the treatment of M. leprae infections in mice. Int. J. Lepr. 44(1976)135-139.
11. SHEPARD, C. C. Statistical analysis of results obtained by two methods for testing drug activity against Mycobacterium leprae. Int. J. Lepr. 50(1982)96-101.
12. SHEPARD, C. C. and CHANG, Y. T. Activity of antituberculosis drugs against Mycobacterium leprae. Int. J. Lepr. 32(1964)260-271.
13. WHO E XPERT C OMMITTEE ON L EPROSY, Sixth report. Geneva: World Health Organization, 1988. Tech. Rep. Ser. 768.
14. WHO STUDY GROUP. Chemotherapy of leprosy for control programmes. Geneva: World Health Organization. 1982. Tech. Rep. Ser. 675.
15. YAWALKAR, S. J. and VISCHER, M. Lamprene(clofazimine) in leprosy. Lepr. Rev. 50(1979)135-144.
1. M.A.;Division of Bacterial and Mycotic Diseases, National Center for Infectious Diseases, Centers for Disease Control. Mail Stop G35, 1600 Clifton Road NE, Atlanta, Georgia 30333. U.S.A.
2. B.S.; Division of Bacterial and Mycotic Diseases, National Center for Infectious Diseases, Centers for Disease Control. Mail Stop G35, 1600 Clifton Road NE, Atlanta, Georgia 30333. U.S.A.
3. Ph.D., Division of Bacterial and Mycotic Diseases, National Center for Infectious Diseases, Centers for Disease Control. Mail Stop G35, 1600 Clifton Road NE, Atlanta, Georgia 30333. U.S.A.
4. Ph.D., Department of Chemistry. University College Dublin, Belficld, Dublin 4, Ireland.
Received for publication on 22 September 1992.
Accepted for publication in revised form on 24 March 1993.