- Volume 59 , Number 1
- Page: 95–101
What next in basic research?*
What's the question?
As we move into the 1990s, it's challenging to try and speculate about areas of basic research which we might expect to provide new insights and developments in the understand ing and control of mycobacterial diseases over the next decade. The central feature of mycobacterial pathogens is their ability to adapt to co-existence within a eukaryotic host, and understand ing of mycobacterial infection at a molecular level relies on elucidation of the steps taken by the host and by the pathogen in stabilizing or destabilizing this interaction. Some of the problems involved in developing a molecular understand ing of the host-parasite interaction are discussed below and are depicted in Figure 1.
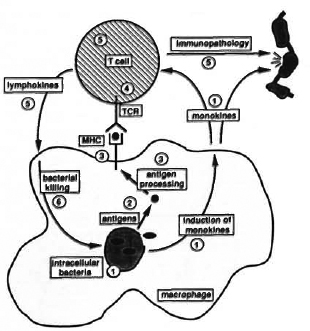
Fig. 1. Molecular mechanisms of mycobacterial infection. A round-trip tour of a range of questions concerning molecular mechanisms of mycobacterial infection is presented: 1 Intracellular bacteria. How do mycobacteria enter cells, and where do they multiply? Do they produce toxins which alter the metabolism of the host cell? Do intracellular mycobacteria induce release of a particular pattern of monokines important in regulation of T-cell activation or tissue damage? 2 Antigens. Which mycobacterial antigens are important in T-cell activation? Secreted proteins? Cell-wall proteins? Heat-shock proteins? 3 Antigen presentation. How are antigens from intracellular mycobacteria processed? Which determinants are presented with which MHC molecules? 4 T-cell recognition. What is the role ofa/fiversusy/dT cells in mycobacterial infection? Are different antigens recognized by different T-cell subsets? 5 T cell function. Arc the T-cell subsets involved in protective immunity functionally distinct from those which cause immunopathology? Can the pattern of lymphokine secretion be used to identify the functional role of T-cell subsets? How docs T-cell function relate to antigen specificity? 6 Bacterial killing. Can activated macrophages kill mycobacteria? If so, what is the mechanism? If not, how arc mycobacteria killed by a protective immune response?
(1) Intracellular mycobacteria. Mycobacteria are generally found inside host cells during infection, but we know little in detail about how they enter the cells or about the conditions which they subsequently experience. There is still uncertainty with regard to their localization in phagosomes, phagolysosomes, or cytoplasm, and we can only guess as to which nutrients are readily available for utilization by intracellular mycobacteria and which may be in limited supply. Phenotypic changes have been reported in infected cells1 - perhaps intracellular mycobacteria produce some subtle form of toxin which subverts host-cell metabolism? Do intracellular mycobacteria induce the release of a particular pattern of monokines from the infected cell which serve to alter the balance of the immune system in favor of the pathogen?
(2) Antigens. We imagine that some components from intracellular mycobacteria become available for antigen processing and subsequent stimulation of host immune responses. Which antigens are important? Perhaps recognition of proteins which are secreted from viable bacilli is the key to protective immunity? 2,3 Or does the adjuvant activity of mycobacterial lipids and carbohydrates confer particular immunological significance on proteins associated with cell-wall structures?4 Alternatively, perhaps the induction of synthesis in response to the intracellular environment- as has been suggested for the heat-shock, or stress, proteins5-is the major factor in determining which antigens are selected for processing.
(3) Antigen processing and presentation. The molecular events involved in antigen processing arc also the subject of speculation. There appear to be two major processing pathways which result in partially degraded antigens being presented on the cell surface bound to a member of the major histocompatibility complex (MHC). 6 Proteins originating from inside the cell generally follow a pathway resulting in presentation with a class I MHC molecule, with external proteins being presented with a class II molecule. Presumably therefore, the intracellular location of particular mycobacterial antigens-i. e., whether or not they escape from phagocytic vesicles-will have some bearing on their immune recognition. In addition to the division between class I and class II MHC molecules, difFercnt haplotypes within each MHC class have marked différences in their capacity to bind and present individual antigenic determinants. Thus, from one antigenic molecule several distinct antigenic determinants can be selected for presentation by different MHC molecules. Associations which have been reported between the incidence of different forms of leprosy and particular MHC haplotypes do indicate a role for such MHC selection in determining the outcome of mycobacterial infection. 7 The question of which antigen? is then compounded by consideration of which determinant? and which MHC molecule? In addition to presentation by infected cells, antigens released from killed bacteria will also be available for uptake by adjacent uninfected antigen-presenting cells, including dendritic cells and B lymphocytes. Docs antigen presentation by infected cells and by uninfected cells provide different signals to the immune system?
(4) T-cell recognition. The cell surface antigen-MHC complex is available for recognition by the antigen-specific receptor on a T lymphocyte. This results in T cell activation-the key trigger for cell-mediated immune responses. The T-cell receptor is a dimcric protein, structurally related to an antibody molecule, consisting of either α and β subunits or γ and δ subunits. Attention has focused principally on T cells with an α/β receptor, but recent reports demonstrating that a high proportion of γ/δ T cells are present in leprosy lesions 8 and are stimulated by mycobacterial antigens 9 serve to emphasize the importance of the question of which receptor is being engaged in mycobacterial infection?
(5) T-cell function: protection and pathology. The next piece of the puzzle concerns the function of the activated T cells. The leprosy spectrum provides a striking illustration to the fact that T-cell activation in mycobacterial disease contributes both to the elimination of the bacilli and to tissue destruction and pathology. Are different subsets of T cells involved in these two aspects? Or do disease and resistance represent a difference in function or regulation of the same set of T cells? T-cell function is thought to be expressed in the form of release of soluble lymphokines capable of triggering metabolic responses in adjacent cells, and by direct cell-cell interactions which can result, for example, in killing of a target cell by cytotoxic T lymphocytes. What functions are involved in mediating protection and pathology? Is the function of a particular T cell phenotypically stable? Or does the profile of secreted lymphokines, for example, reflect the environment of the cell at any one time?
(6) Killing of mycobacteria. It is assumed that a major aspect of T-cell function in protective immunity is to convert macrophages into a mctabolically active form in which they are capable of killing ingested mycobacteria. It has proved extremely difficult to obtain experimental evidence for this model, however, and the mechanisms which macrophages use to eliminate intracellular mycobacteria remain the subject of speculation. How might such killing be brought about? This question brings us back to the aspects discussed above regarding our lack of knowledge about intracellular mycobacteria. Perhaps there are nutrients or trace elements which arc of particular importance to the intracellular bacilli and, by ensuring that they are not available, the macrophage might just slowly starve the mycobacteria to death? This would be consistent with the ability of the immune system to restrict mycobacterial growthi n vivo and the difficulty of reproducing the phenomenon in short-term in vitro cultures.
In setting out this list of questions, I have tried to make two things clear. Firstly, it is very easy to formulate a large number of questions concerning molecular mechanisms of mycobacterial infection. Secondly, however, I have tried to indicate the way in which these questions are inter-related in the almost circular form depicted in Figure 1. If we could identify the antigens from viable bacilli which arc available for processing, for example, then this would help us to identify the T cells which might be important in regulating the infection. If we were to identify lymphokines that mediate protective responses then, by analyzing their effect on macrophage function, we might reveal the mechanisms involved in killing of intracellular mycobacteria. Or, vice versa, if we understood the mechanism of killing, then we could search for lymphocytes capable of inducing such functions. In other words, rather than trying to say that any of the questions listed above in itself represents the key to understand ing mycobacterial disease, in reality, acquiring knowledge in any of these subjects would have a knock-on effect which might lead to breakthroughs in other questions. Thus, rather than imposing a hierarchy of importance, we should take a pragmatic attitude and be grateful for progress in any of the areas listed above. In which areas can we expect some progress in the next few years? I would like to discuss this under three headings of molecular biology, immunology and diagnostics.
What is the answer?
Molecular biology. In his younger days, a now distinguished molecular biologist castigated an audience of lcprologists at Carville, Louisiana, U. S. A., with the taunt that they knew nothing about Mycobacterium leprae. He went on to qualify the statement by saying that he was making a comparison with Escherichia coliand, much to his credit, has spent the intervening years trying to remedy the situation. I think that the gulf between M. leprae and E. coli- or rather, the possibilities of bridging it-holds the most exciting potential for fundamental new insights into mycobacterial disease in the 1990s. Molecular geneticists have spent decades in understand ing E. coli. Now that the tools of molecular genetics arc beginning to prise open the mycobacterial pathogens, 10-13 we can pose an exciting challenge to basic scientists. On the one hand, we have the vast amount of detailed knowledge of E. coli genetics; on the other, the blank page of mycobacterial genetics. How should we start to fill in our knowledge of mycobacteria? Surely not by reproducing all that has been done with E. coli. Can we use that knowledge, however, to identify those aspects of mycobacterial genetics which are most relevant to their pathogenicity? It is important that the new developments in molecular genetics are not simply used as an excuse for the straightforward repetition of oldE. coliexperiments with another organism, but rather that they are used to lend a new perspective to the complex and intellectually demand ing questions concerned with mechanisms of mycobacterial infection.
Perhaps growth rate is the most obvious starting point in comparing mycobacteria and E. coli. Why does M. tuberculosis grow so slowly and M. leprae-in vitro -not at all? I can't believe that this is because mycobacteria are slow witted or defective in some way. It seems much more likely that this characteristic has been selected in order to facilitate the interaction with their eukaryotic hosts and is probably crucial to their success as parasites. The bulk of our knowledge of prokaryotic gene regulation is derived from study of E. colicells with a division time of around 20 minutes-are there any fundamental differences in a bacterium which divides only once in 24 hours, or once every week or so? In addition to academic interest, investigation of this type of question may provide novel perspectives relevant to disease control. Most adult tuberculosis results from reactivation of apparently dormant infection. Perhaps a fundamental study of gene regulation will allow us to define some state of mycobacterial dormancy and might identify mechanisms involved in switching between dormancy and active multiplication. It can be imagined that mycobacteria will display different levels of drug sensitivity at different stages of their growth cycle, and insights derived from the study of such basic phenomena might therefore point the way to improved chemotherapy regimens allowing more rapid elimination of persister organisms and reduction in the length of treatment.
A second aspect of gene regulation which is now open to study is the question of mycobacterial adaptation to the host-parasite interactionin vivo. A central feature of bacterial pathogenicity research over the last few years has been the discovery that expression of genes encoding components involved in interactions with host cells (including adhesins and toxins-the classical virulence factors) is induced in response to environmental signals encountered during infection. 14 Is it also the case for mycobacteria that factors which are of particular importance for intracellular survival are induced following phagocytosis? Evidence from other bacterial pathogens suggests that heat-shock proteins are included among the proteins induced by entry into host macrophages,15 and analysis of the regulation of these genes in mycobacteria might lead to the identification of other co-regulated genes encoding factors important for intracellular growth. Analysis of such factors should provide information about the conditions experienced by intracellular mycobacteria. We might anticipate, for example, finding genes encoding components of transport systems for nutrients which are particularly important forin vivosurvival. Studying gene regulation in mycobacteria would then be an indirect monitor of conditions within the infected host cell, and might allow us to identify host cell metabolic features which are conducive or inhibitory to mycobacterial survival. Proteins which are induced during intracellular growth may also be of particular interest to the immune system. Lymphocytes directed to such antigens might specifically recognize cells harboring live organisms, and antibodies to induced proteins may distinguish progressive infection from subclinical infection in the absence of bacterial growth.
Molecular genetics also offers the possibility of modifying mycobacteria. For M. leprae, it may be possible to introduce new genes, or to inactive control mechanisms, in order to create mutant strains adapted fori n vitroculture. For cultivable mycobacteria, it will be possible to follow procedures adopted with other pathogens of deleting specific genes in order to evaluate the role of individual molecules, or regulatory pathways, in the overall process of infection. 16 In addition, the creation of rationally attenuated mutant strains can be envisaged, with genetic alterations designed to destroy pathogenicity while preserving immunogenicity. Generation of such strains in the 1990s would represent a second step along the pathway initiated by Calmette and Gucrin in the early decades of the century. Attempts to modify the BCG vaccine itself by introduction of genes encoding protective antigens from other pathogens are already underway,10-12 and this approach could be applied to development of novel antimycobacterial vaccines if the appropriate antigens are identified.
Immunology. The normal immune response of most individuals living in leprosyendemic areas seems to be sufficient to provide resistance to clinical disease, and the concept of using a vaccine to enhance or modify immunity in the remaining susceptible portion of the population is an attractive prospect. The goal of rational vaccine development was enthusiastically embraced during the 1980s following dramatic breakthroughs in antigen availability. How far have we progressed in an understand ing of the molecular mechanisms of mycobacterial immunity, and what are the prospects for a genetically engineered subunit vaccine in the next decade? The answer to this question depends on our view of the nature of protective immunity to mycobacteria.
The simplest model of the immune response to mycobacteria would envisage a situation in which disease occurs when the immune system is switched off, and disease is prevented when the system is on (Fig. 2A). Off might represent failure to recognize the infection, or active suppression, and would be illustrated classically by lepromatous leprosy. The pathology associated with tuberculoid leprosy could be explained by a delayed switch on causing an over-reaction by the immune system subsequent to the establishment of the infection. On the basis of this model, one would look for a vaccine containing sufficient antigenic determinants to prime a broad spectrum of T cells, and one might feel reasonably optimistic that such a preparation could indeed be obtained.
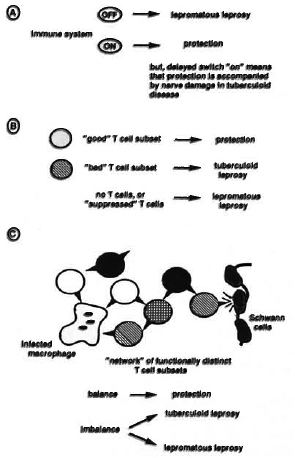
Fig. 2. Models for cellular immunity in leprosy. Three schematic representations of possible cellular immune responses in the leprosy spectrum arc presented, showing a simple on-off' system in A, a more complex model involving protective and pathological T-cell subsets in B, and a balanced network of interacting lymphocytes in model C.
A more elaborate view of the immune system would envisage the occurrence of separate protective and pathogenic aspects to immune activation. Perhaps the vigorous T-cell activation manifest in most forms of mycobacterial disease-including tuberculosis and tuberculoid leprosy -is of a different nature from that responsible for protective immunity? One might then consider the possibility of protective and pathogenic T-cell subsets as depicted in Figure 2B. On the basis of this model, we could think of a vaccine in terms of specifically inducing functionally distinct T-cell subsets using different antigenic stimuli. The fact that we have little idea as to the nature of protective T cells in mycobacterial disease, and that there is a lack of certainty in the equation of antigen specificity with T-cell function, may make one less optimistic about the prospects for rational vaccine development along these lines. If pathogenic T-cell subsets are involved in tissue damage, perhaps we should also consider the possibility of developing vaccines, or therapies, based on the idea of specifically inhibiting certain forms of T-cell activation-by analogy with strategies designed to combat autoimmune diseases?
Further sophistication of our model of cellular immunity would include consideration of a possible network of interacting lymphocytes which express a variety of functional activities and with complex patterns of communication with each other in addition to response to foreign antigens (Fig. 2C). In this model, protective immunity would arise from an appropriate balance of immune activities-possibly involving both lysis and activation of phagocytic cells 17- while disease could result from the same immune functions activated in an unbalanced or inappropriately regulated fashion. The different clinical symptoms of the leprosy spectrum would then be seen as a manifestation of the immune system being knocked out of balance in different directions. Designing a vaccine to set up, or to restore, a complex balance in the immune system would seem to present a rather formidable challenge, with attenuated whole cell vaccines perhaps seeming more attractive than defined subunit preparations.
In discussing the prospects for progress in molecular biology, there is an opportunity to contrast mycobacteria with well-defined models derived from study of other bacteria. The difficulties encountered in trying to uncover molecular mechanisms of mycobacterial immunity, on the other hand, are shared by the whole field of cellular immunology. Elucidation of fundamental mechanisms of cellular immunity is a goal which is actively pursued by a substantial section of the basic research community and, in order to ensure progress in mycobacterial immunity, it is important that the problems we face arc expressed in a form accessible to active workers in this basic field. This allows rapid application of new ideas arising from basic research, and also has the effect of increasing general awareness and interest in the problems associated with mycobacterial disease. Persuading immunologists to switch from sperm-whale myoglobin to a mycobacterial heat-shock protein as a model system is a useful step in this direction.
Diagnostics. Leprosy has traditionally been diagnosed on the basis of a) detection of the organism by skin smear, and b) detection of the disease by clinical examination for skin and nerve lesions, and by histopathology. The input from molecular scientists in the field of diagnostics has been directed essentially toward detection of the organism. Perhaps there is scope for development of more disease-oriented diagnostic tests in the 1990s?
Identification of antigenic determinants specific to M. leprae in the early 1980s prompted renewed efforts to develop sérodiagnostic tests for leprosy. 18 Circulating antibodies were used to detect M. leprae and conscientiously reported back that a lot of M. leprae is present in multibacillary disease, and that a little M. leprae is present in paucibacillary disease and in a proportion of contacts presumably exposed to some form of subclinical infection. Perhaps the next generation of diagnostic tests will be able to probe for immune responses which are specific to progressive, clinically relevant infection? As discussed above, molecular genetics may identify antigens induced during intracellular growth which would represent useful new serological targets. Advances in understand ing of molecular mechanisms of cellular immunity might allow the distinction of protection from pathological forms of immune activation, and could point the way toward development of tests for immune phenomena, specifically associated either with resistance or with disease.
In terms of sensitivity, both serological tests and the traditional smear evaluation arc now undoubtedly surpassed by the remarkable developments of molecular biology in the form of the polymerase chain reaction (PCR). 19,20 PCR provides an exquisitely sensitive and specific tool for detecting perhaps as little as a single mycobacterial genome, and it is hoped that further development to reduce the technical difficulties and cost of the assay will result in its becoming available in routine diagnostic laboratories. It will be fascinating to see how widespread M. leprae is within exposed populations. Will there be a significant proportion of individuals harboring a few apparently harmless bacilli as predicted by immunodiagnostic assays? Or will the presence of the organism actually correlate with the presence of clinical disease? It is anticipated that the use of gene probes and PCR techniques will have much to reveal about the epidemiology of mycobacterial infection over the next decade.
Returning to the idea of detection of the disease process, one might anticipate that further effort to understand neuropathology at a molecular level could lead to development of early tests for nerve damage in leprosy. Detection of autoantibodies to nerve components is under active investigation,2 1 and perhaps breakdown products from damaged nerves could themselves be successfully identified in serum samples? Another class of molecules related to the disease process which may be useful diagnostic targets are the soluble mediators of the immune response. Serological detection of tumor necrosis factor is one example, 22 and perhaps, as we refine our understand ing of immune mechanisms, particular lymphokine patterns will be seen to distinguish protective and pathogenic forms of immune activation.
Concluding remarks
The most exciting aspect of fundamental research is not just in finding a solution to a defined problem, but in the possibility that, from some novel perspective, the problem itself will no longer exist. In an era when we arc seeing the ground rules of mycobacterial disease control undergo dramatic changes in HIV-infected populations, it is more than ever important that, along with pursuit of established goals-implementation of multidrug therapy, testing of vaccine cand idates, and assessment of existing diagnostic tests-we continue to try to expand our basic understand ing of molecular mechanisms in the hope of uncovering new concepts for application to the control and prevention of mycobacterial disease.
- Douglas B. Young, Ph. D.
MRC Tuberculosis and Related Infections Unit
Royal Postgraduate Medical School
Hammersmith Hospital
Du Cane Road
London W12 OHS, England
1. Sibley, L. D. and Krahcnbuhl, J. L. Induction of unresponsiveness to gamma interferon in macrophages infected with Mycobacterium leprae. Infect. Immun. 56(1988)1912-1919.
2. Rook, G. A. W., Steele, J., Barnass, S., Mace, J. and Stanford, J. L. Responsiveness to live M. tuberculosis ,and common antigens, of sonicate stimulated T cell lines from normal donors. Clin. Exp. Immunol. 63(1986)105-110.
3. Orme, I. M. The kinetics of emergence and loss of mediator T lymphocytes acquired in response to infection with Mycobacterium tuberculosis. J. Immunol. 138(1987)293-298.
4. Melancon-Kaplan, J., Hunter, S. W., McNeil, M., Stewart, C, Modlin, R. L., Rca, T. H., Convit, J., Salgame, P., Mehra, V., Bloom, B. R. and Brennan, P. J. Immunological significance of Mycobacterium leprae cell walls. Proc. Natl. Acad. Sci. U. S. A. 85(1988)1917-1921.
5. Young, D., Lathigra, R., Hendrix, R., Sweetser, D. and Young, R. A. Stress proteins are immune targets in leprosy and tuberculosis. Proc. Natl. Acad. Sci. U. S. A. 85(1988)4267-4270.
6. Germain, R. N. The ins and outs of antigen processing. Nature 322(1986)687-689.
7. de Vries, R. R. P., Ottenhoff, T. H. M. and van Schooten, W. C. A. Human leukocyte antigens (HLA) and mycobacterial disease. Springer Semin. Immunopathol. 10(1988)305-318.
8. Modlin, R. L., Pirmez, C, Hofman, F. M., Torigian, V., Uyemura, K., Rea, T. H., Bloom, B. R. and Brenner, M. B. Lymphocytes bearing antigen-specific γδ T-cell receptors accumulate in human infectious disease lesions. Nature 339(1989)544-548.
9. Bom, W., Happ, M. P., Dallas, A., Reardon, C, Kubo, R., Shinnick, T., Brennan, P. and O'Brien, R. Recognition of heat shock protein and γδ cell function. Immunol. Today 11(1990)40-43.
10. Jacobs, W. R., Tuckman, R. and Bloom, B. R. Introduction of foreign DNA into mycobacteria using a shuttle phasmid. Nature 327(1987)532-535.
11. Snapper, S. B., Lugosi, L., Jekkel, A., Melton, R. E., Kieser, T., Bloom, B. R. and Jacobs, W. R. Lysogcny and transformation in mycobacteria: stable expression of foreign genes. Proc. Natl. Acad. Sci. U. S. A. 85(1988)6987-6991.
12. Husson, R. N., James, B. E. and Young, R. A. Gene replacement and expression of foreign DNA in mycobacteria. J. Bacterid. 172(1990)519-524.
13. Martin, C., Timm, J., Rauzier, J., Gomez-Lus, R., Davies, J. and Gicquel, B. Transposition of an antibiotic resistance element in mycobacteria. Nature 345(1990)739-743.
14. Miller, J. F., Mekalanos, J. J. and Falkow, S. Coordinate regulation and sensory transduction in the control of bacterial virulence. Science 243(1989)916-922.
15. Buchmeicr, N. A. and Heffron, F. Induction ofSalmonellastress proteins upon infection of macrophages. Proc. Natl. Acad. Sci. U. S. A. 248(1990)730-732.
16. Finlay, B. B. and Falkow, S. Common themes in microbial pathogenicity. Microbiol. Rev. 53(1989)210-230.
17. Kaufmann, S. H. E. CD8+ T lymphocytes in intracellular microbial infections. Immunol. Today 9(1988)168-174.
18. Serological tests for leprosy (Editorial). Lancet 1(1986)533-535.
19. Hancc, A. J., Grand champ, B., Levy-Frebault, V., Lecossier, D., Rauzier, J., Bocart, D. and Gicquel, B. Detection and identification of mycobacteria by amplification of mycobacterial DNA. Mol. Microbiol. 3(1989)843-849.
20. Hartskeerl, R. A., de Wit, M. Y. L. and Klatser, P. R. Polymerase chain reaction for the detection ofMycobacterium leprae. J. Gen. Microbiol. 135(1989)2357-2364.
21. Itty, B. M., Mukcrjce, R. and Talwar, G. P. An enzyme immunoassay (EIA) based on antibodies against human nerve antigen for diagnosis of all categories of leprosy patients. (Abstract). Int. J. Lepr. 57 Suppl. (1989)304.
22. Grau, G. E., Roux-Lombard, P., Gysler, C., Lambert, C., Lambert, P. H., Dayer, J. M. and Guellevin, L. Serum cytokine changes in systemic vasculitis. Immunology 68(1989)196-198.
*Based on a talk Summing Up: What Next?, presented at the Sixth Joint Meeting of the IMMLEP, IMMTUB and THELEP Steering Committees in Geneva, Switzerland, April 1990.