- Volume 58 , Number 2
- Page: 365–75
An ordinary mortal's guide to the molecular biology of mycobacteria*
Editorial opinions expressed are those of the writers.
XIII LEPROSY CONGRESS STATE-OF-THE-ART LECTURE
We are pleased to have the opportunity of publishing the full texts of the state-of-the-art lectures presented at the XIII International Leprosy Congress at The Hague, The Netherlands, 11-17 September 1988. This lecture completes the series.
In the beginning was the Word and the Word was with God, and the Word was God."
- John 1. i.
There is perhaps no more vivid way to convey the sense of profundity, centrality, power and wonderment of molecular biology in the context of biology than to recall the opening words of the Book of John. The choice of metaphors is not at all intended to be blasphemous; indeed, in a curious way, I think that these words may be interpreted to pertain more literally than one might anticipate. The most fundamental tenet of the molecular genetic approach of the new biology is the "central dogma," which holds that:
i)The genetic code is written in molecules of dna (deoxyribonucleic acid); and
ii)Genes encode the information to make proteins, by a process of
![]()
To some it will be surprising that in the modern conception of the molecular basis of heredity one finds the terms and concepts for " central dogma," genetic code, transcription, translation, and library -phrases that are used every day by molecular biologists. That resonance between the simplest of ideas and the enormous complexity of mechanisms is truly part of the fascination of molecular biology.
The basic challenge of molecular biology is to explain how the complexity of an individual, the diversity of individuals within populations, and the evolution of species within the biosphere can be described in chemical and physical terms, and how the potential for this diversity of life, present initially within a single cell, can be realized. The major evolutionary problem that had to be solved is how to " encode" specific information for heredity in one or more molecules, and how to ensure that those carrying genetic information can be faithfully replicated in each cell division. DNA represents the solution of that problem. The key feature of DNA molecules is that they have the ability precisely to replicate themselves, as cells and chromosomes divide. This is possible because DNA consists of two, complementary, intertwined molecules (known as a heteroduplex) bound to one another in the familiar (α-helix. Molecular biologists have learned that large DNA molecules encode the information, called genes, for all of the complex enzymes and chemical substances that pattern the construction of cells in all forms of life, and that all forms of life use basically the same code, i.e., the same language, to create their widely differing proteins. Rather than 26 letters of the Roman alphabet, the infinitely richer and more variegated genetic code uses only four: A, T, C, G. These letters stand for nucleic acid bases, simple chemical compounds, that are chemically linked together to form very large molecules that make up the deoxyribonucleic acid, or DNA. A stands for adenine; T for thymidine; G for guanine and C for cytidine. The choice of the four bases in DNA permits replication through an extraordinarily sophisticated and subtle mechanism. The four bases have " chemical complementarity," that is, in three-dimensional structures of DNA, A can bind to T, G can bind to C (Fig. 1). The result of this, when these long polymers of DNA chains are in solution, is that they form complementary structures of two long strands, with " base pairing." When one chain unfolds, and appropriate enzymes are present, free bases complementary to the original strands can be bound, e.g., a free A pairs with T and a free G with C, and then are chemically linked or zippered to produce an exact copy in reverse orientation of the original DNA strand. This, then, is a fully self-replicating system which preserves precisely the fidelity of the original chemical genetic information.
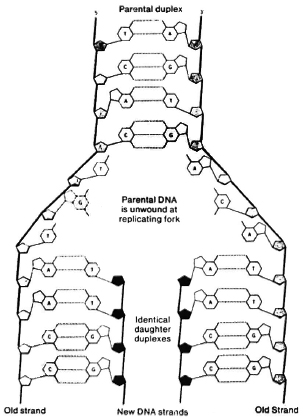
Fig. 1. Base pairing.
Perhaps because of the evolutionary need to protect the key molecule containing the genetic code, DNA is not itself used directly to manufacture proteins, but rather is " transcribed" into another set of smaller nucleic acid molecules, known as ribonucleic acid or RNA, each of which can code for one or a small number of proteins or polypeptides. Three of the bases used to construct DNA are also used in RNA; one, " U" for uracil, is unique to RNA. The RNA bases bind by similar complementarity to stretches of DNA genes encoding particular proteins, and it is these structures complementary to DNA that actually provide the imprint for producing the intended protein. It is the RNA molecules that bind to the protein-synthesizing machinery of cells and are " translated" into specific protein molecules.
The genetic code
The question then is how do four simple chemical molecules permit the precise specification of protein molecules, which are made of up to 22 amino acids, in precisely the correct order for each protein? The solution has evolved in the form of a " triplet code," i.e., three nucleic acids are required to precisely specify a single amino acid. Each triplet of three nucleic acids that encodes a single amino acid is called a " codon." The various codons make up a dictionary of amino acids used in proteins (Fig. 2). The universality of the code means that essentially all bacteria, viruses, plants, protozoa, lower animals and man use the same three nucleic acids to encode the same amino acid in a given position in a protein. For example, the codon ATG specifies the amino acid methionine. DNA molecules are millions of base-pairs long. For example, the entire genome of Mycobacterium leprae has about 3 x 106 base pairs, while the human genome represents about 8 x 109 base pairs, molecules long enough from a single cell to spread out 4 cm. Yet individual proteins are much shorter. To solve that potential problem, the genetic " code" even has " punctuation," i.e., triplet codons that specify the termination of a protein sequence, just as a period terminates a sentence.
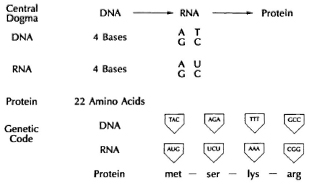
Fig. 2. The genetic code.
Chance and necessity
If the genetic code is universal and the triplet code specifies each of the amino acids across all of evolution, how can the diversity of life be accounted for? There are a number of mechanisms for a process of genetic " mutation." Basically, all studies since the time of Darwin have indicated that genetic change, known as mutation, occurs continuously and randomly at a low frequency, perhaps one mutation in any specific character or trait per million cell generations. For example, M. leprae or M. tuberculosis would be expected to develop one random mutant resistant to any individual antibiotic in every million organisms.
How docs mutation work? In order to change a biological character or, more precisely, a " gene product," i.e., a protein, one has to change at least one amino acid. The simplest way to do that is known as a point mutation or " misprint," in which one of the three nucleic-acid bases in the original triplet codon is simply chemically changed or substituted by another such that, in the dictionary of the genetic code, a different amino acid is inserted at that point in the protein. However, as shown Figure 3, if one inserts an extra base in a particular position in DNA, one alters not only the triplet code for one particular amino acid by a single base but shifts all the letters in the code following that insertion, making a whole new set of triplet codons. This is called a " frame shift." The consequences are that not only is there a single amino acid changed at the site of insertion, but every subsequent codon is completely different. Thus, by the insertion of a single base or, obviously, the deletion of a single base, the genetic code is dramatically altered for that protein. This permits production of completely different sequences of amino acids in large polypeptides.
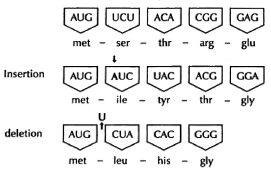
Fig. 3. Mutation.
The consequences of the process of random mutation are profound. While it provides subtle modifications of proteins as the result of point mutations for single amino acids, it allows for the production of entirely new proteins by the insertional or deletional mechanism of mutagenesis. It should be obvious that the vast majority of such mutations that occur are deleterious. A " frame shift" that changes all the amino acids in the polypeptide chain after the insertion of one nucleic acid base generally produces a protein that cannot carry out its useful biological function, and such mutated proteins are usually rapidly degraded. Because many of these changes would have occurred in molecules vital for the cell, mutations in essential proteins most often lead to cells that cannot survive. Essentially, the only circumstance in which mutations are able to persist are those in which a change is produced in a particular protein that is selected for by the environment to permit greater survival or growth of that cell or organism than the parent from which it originally derived. This is the fundamental Darwinian basis of heredity. Natural selection acts on products of individual genes within individuals, and those with some advantage are selected for.
How do we identify genes, i.e., sequences of bases in DNA, that encode a particular protein or function? In bacteria, often the first step is to create and to select for a mutation that alters the particular function controlled by the gene we are interested in. For example, if we want to study the synthesis of nucleic acids, and we assume it takes multiple enzymes to make each of the nucleic acid bases, one can look for mutants which are unable to grow in ordinary medium but are able to grow in medium supplemented with a particular base, such as " U," required for RNA synthesis (Fig. 4). In the absence of RNA, cells fail to make protein and, in the absence of protein, they fail to replicate and make DNA. Thus, wc can look for a mutant that fails to grow in regular medium because enzyme " C" is mutant, but which will grow in medium supplemented with uracil. An organism that has a mutation that inhibits growth, but which can be grown by the addition of some chemical substance to its environment, is called an " auxotroph." Now, molecular biologists would obtain pieces of DNA from the original parental strain (called " wild type" ) from which the mutant was derived and which contained the correct gene for producing U. They would then add these DNA pieces to the mutant. To transfer DNA effectively between organisms, they need a vector, a form of DNA that can both transfer DNA between cells and also replicate in the mutant cell. If any of the pieces inserted into the vector contain an intact gene that carries out the function of the mutant gene " C," the organisms will now grow in minimal medium without the addition of uracil. We would say that the defect is " complemented," and the cells can now complete their synthetic pathway. It is by this simple procedure, of producing mutations and then identifying DNA sequences that complement them, that most of the molecular genetics of simple organisms such as Escherichia coli has been described. As will be seen later, we would hope to use this concept of genetic complementation to devise a strategy to permit cultivation of M. leprae in the test tube.
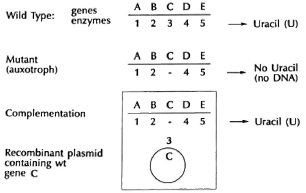
Fig. 4. Complementation.
Revolution beyond evolution
Because DNA is a self-replicating molecule, molecular biology permits scientists to do things that are not possible in nature. For example, while M. leprae docs not grow in culture, DNA from M. leprae can be grown in infinite amounts. If M. leprae DNA, for example, is put into a suitable vector, it is possible to introduce the vector into E. coli and have it replicate in its vicarious host millions of copies of the original M. leprae DNA, each with the identical nucleic acid sequence as the original piece of DNA. This is the process of " cloning," and the manipulations that allow it to be carried out are commonly known as " genetic engineering." The molecular biological revolution makes it possible to create in a week new molecules that would take mil-lenia to be produced in the time-frame of evolution, that have properties and activities which exceed any that have naturally evolved. If one stably introduces a gene in the right form which allows it to replicate, foreign proteins can be " expressed" in the host cell. Because the foreign DNA is " re-combined" with the DNA of the host cell, this represents the basis for " recombinant DNA" technology.
Manipulating mycobacteria
One of the aims of our work is to develop BCG into a multivaccine vehicle capable of immunizing an individual against several antigens from different pathogens simultaneously. In order to do that we must be able to develop a recombinant BCG, and to introduce and to express genes, i.e., carry out transcription, translation and synthesis of proteins, for foreign antigens in BCG.
Therefore, vectors have had to be constructed which have at least three requisites in order to permit their own survival and the expression of a foreign gene (Fig. 5). There are basically two types of vectors that are used to exchange DNA between cells. One form is the DNA from viruses that infect bacteria, called phages (Fig. 6). While all phages can introduce DNA into bacteria by infection, most grow and ultimately lyse the bacterial host, releasing many thousands of phage particles. Of particular interest for our purposes are a type of phages called temperate, which do not lyse the cell but integrate into the host-cell chromosome and divide when the cell divides. The most commonly used vector consists generally of circular pieces of DNA called plasmids that replicate in the cytoplasm (Fig. 7).
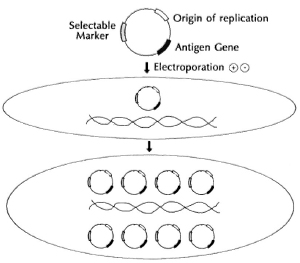
Fig. 5. Vectors, plasmid transformation.
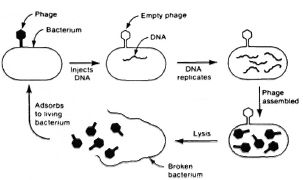
Fig. 6. Phages.
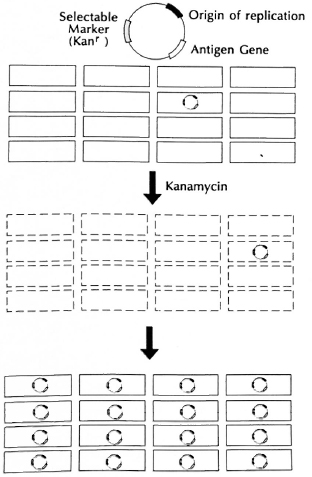
Fig. 7. Genetic selection.
In addition to the condition that the vector not kill the host cells, there are basically three requisites of a useful vector for our purposes. First, the vectors must be capable of replicating in mycobacteria, and thus they need a segment of DNA which functions as a " replicon," or origin of replication. Second, they must, of course, carry the antigen genes we wish to express. And third, they need a " selectable marker." The introduction and expression of foreign DNA by a vector in a stable way is known as the process of " genetic transformation." In most bacteria, that is a relatively inefficient process since it requires moving highly charged, large DNA molecules across the cell wall and cell membrane, having it survive exposure to nucleases, enzymes that degrade nucleic acids like DNA and RNA. Only one in a thousand to one in ten thousand bacilli actually take up foreign plasmid DNA under usual conditions. In order to be able to obtain precisely those organisms which have taken up the foreign DNA and may thus express the foreign antigen, one needs a " genetic selection" (Fig. 7) that will permit only " transformants" to survive and will kill or allow to die all the other organisms that have not taken up the appropriate DNA. The usual procedure by which this is carried out in bacterial genetics is to introduce a " selectable genetic marker" on the introduced DNA. For experimental work, that marker is usually a gene which provides resistance to a particular antibiotic that normally kills the species of organism under study. Thus, to transform E. coli, one often introduces a circular piece of DNA, called a plasmid, containing an origin of replication, the gene of interest and a selectable marker gene for resistance to an antibiotic, let us say, a gene encoding kanamycin resistance. If only one cell in a million is able to take up this DNA, we nevertheless have the potential to allow that one organism to grow up by selecting for it in medium containing kanamycin. Since kanamycin will kill all of the organisms not expressing that gene, we can basically select for the growth of one organism in a million. If that plasmid which is being selected for also contains the gene for a foreign antigen, then we will be simultaneously selecting for the antigen gene when we select for survival for organisms containing the selectable marker or antibiotic resistance gene.
a shuttle strategy
Because mycobacteria such as BCG grow so extraordinarily slowly in culture (e.g., a colony of E. coli can be seen on a plate in 8 hours, but an equivalent sized colony of BCG would require 24 days). Bill Jacobs devised the key strategy in our laboratory for manipulating DNA in a vicarious host that is easy to work with, such as E. coli, and then " shuttling" it into mycobacteria. Initially, there were formidable problems in introducing any foreign DNA into mycobacteria because of the very refractory lipid-containing cell wall. The first breakthrough was made by Jacobs, who reasoned that the one agent that one knew with certainty could introduce DNA into mycobacteria was " mycobacteriophages." Phages are viruses that infect bacteria, and have long been used in the typing of tubercle bacilli and other mycobacterial species from clinical isolates. He created a novel vector called a " shuttle phasmid," which is a mycobacteriophage that carries within its DNA a plasmid capable of replicating and growing in E. coli. Thus, his first vector was able to grow as a plasmid in E. coli where genes could be inserted and manipulated, and then to grow in mycobacteria as a phage. With these plasmids, Jacobs introduced foreign genes for the first time into mycobacteria, including BCG. One of the important consequences of this strategy was that he was able to identify the first, useful, selectable genetic marker for the mycobacteria, namely, a gene encoding resistance to kanamycin.
Because many plasmids can replicate in many more copies in bacteria than temperate phages, and thus produce more recombinant proteins, in collaboration with T. Kieser, A. Jekkel and colleagues in Norwich, Scott Snapper and Bill Jacobs created a shuttle plasmid which has the ability to exchange or " shuttle" DNA between E. coli and mycobacteria, and demonstrated that it worked (Fig. 8). Their reasoning was that M. fortuititm had previously been reported 1 to contain a natural plasmid called pAL5000, albeit one that was not known to have any function or selectable markers, and that the very existence of a plasmid associated with the M. fortuitum species indicated that, whatever else it lacked, the plasmid must have the ability to replicate in mycobacteria, i.e., possess a replicon. Consequently, a hybrid plasmid was made in which an E. coli plasmid, with all of the appropriate antibiotic resistance selectable markers as well as an E. coli replicon, was genetically engineered into the plasmid from M. fortuitum to create a hybrid plasmid that has a mycobacterial origin of replication as well as antibiotic selectable markers. This plasmid was used successfully to transform M. smegmatis to resistance to kanamycin.2
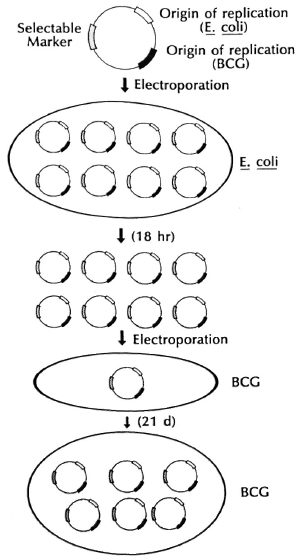
Fig. 8. Shuttle plasmids.
Thus, all of the genetic manipulations, introduction of selectable markers, cloning of foreign antigen genes, growing in large amounts, can be done in E. coli readily. They dramatically improved the frequency of transformation, i.e., the number of organisms expressing foreign genes, through the use of a new technique called " electro-poration," in which the highly charged DNA and cell surface are exposed to very high electrical potentials for very short periods of time. Apparently damage is done to the membrane by the electrical potential and, because of its charge, DNA is forced into the cell. The shuttle plasmids they constructed have replicons both for E. coli and for mycobacteria, as well as selectable antibiotic resistance markers that permitted selection of the shuttle plasm id vector both in E. coli and mycobacteria. It is particularly gratifying that Dr. L. Lugosi, working in our laboratory and one of the world's experts on BCG, has been able to introduce foreign DNA on these shuttle plasmids and to select for transformants expressing foreign genes in both E. coli and in mycobacteria, including BCG vaccine substrains (Fig. 9).
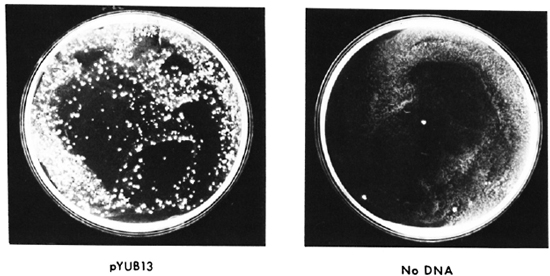
Fig. 9. Transformation of BCG.
Looking at libraries
If we are to construct a multivaccine vehicle in BCG, how are we to obtain the genes for protective antigens, for example, from M. leprae, to insert into our shuttle vectors? The strategy that has been most generally useful in this regard was developed by Richard Young and Ron Davis through the use of a special E. coli bacteriophage, called lambda gt11 (λgt11). The concept was to make a " library" of all the genetic information in the chromosome of M. leprae or M. tuberculosis in a simple bacterial virus that infects E. coli (Fig. 10). To make a genetic library, in essence, the DNA of the mycobacterium or any organism is broken into many small pieces and each piece is inserted into the DNA of a separate bacteriophage. All the genes of the organism are thus divided into and represented by a million bacteriophages. Since λgt11 infects and grows in E. coli, literally billions of copies of any bacteriophage can be grown up overnight, if we wish. The phages are constructed in such a way that the foreign genes are inserted in the middle of the gene encoding an E. coli enzyme, beta-galactosid-ase, that loses its ability to turn a particular substrate into a blue-colored product when the foreign DNA is present. This enables one easily to select for those infected E. coli containing mycobacterial DNA, since they are white. When these bacteriophages are used to infect E. coli, they produce many, many copies of themselves and, in the process, kill and lyse their E. coli hosts, forming " plaques." Yet, each plaque will contain a small number of mycobacterial proteins encoded by a single piece of mycobacterial DNA. Using antibodies, then, to detect M. leprae antigens, it was possible for R. Young to identify individual plaques that contain bacteriophages with the gene for a single antigen of M. leprae.
Fig. 10. DNA library.
It is by this means that six or seven major antigens of M. leprae and M. tuberculosis have been identified in recombinant λgt11 libraries and the genes for many of those antigens have been obtained and identified. Once the DNA sequence encoding the mycobacterial DNA insert is obtained, it is possible by using the genetic code to predict the amino acid sequence of the protein. Thus, it is possible to guess the amino acid sequence of an antigen even if there is insufficient protein to determine the amino acid sequence of that protein. One of the striking findings of the mycobacterial antigen genes, once their DNA sequence was determined, is that almost half of the major antigens have predicted amino acid sequences very similar to a known class of proteins found in all living organisms, known as heat-shock proteins (hsp) or stress proteins.3 These are proteins that are rapidly produced when organisms are " stressed" by being heated, by being deprived of nutrients, or by being exposed to toxic molecules, such as hydrogen peroxide. Their precise functions are unknown; it seems that they are critically involved in the proper folding of proteins and in transporting proteins across membranes. One interpretation is that bacteria that find themselves in the animal host are " stressed" and produce different antigens than they would make in culture. One serious implication of this result is that heat-shock proteins are highly conserved, that is, similar in structure throughout evolution. Thus, we know that M. leprae hsp65 has about 50% of its amino acids identical to that of E. coli-but also to the human hsp65. Thus, there is a clear antigenic similarity between certain mycobacterial proteins and human counterparts. This has suggested a hit-and-run model for certain autoimmune diseases, for example, rheumatoid arthritis, and possibly other autoimmune diseases. An individual is infected with a mycobacterium and makes an immune response to, among other antigens, the heat-shock proteins. Some of the T cells made to the bacterial antigens then cross-react with the human hsp counterpart found in increased amounts in inflamed tissue. Even after killing the bacterium, the system is able to maintain a chronic autoimmune response. It is an intriguing hypothesis that is currently being actively pursued. It also means that candidate antigens for protective vaccines must be examined critically for their ability to generate autoreactivity.
Once protective recombinant antigens are identified, it is then a relatively easy matter to excise the piece of mycobacterial DNA encoding the M. leprae antigen gene from the λgt11 expression vector and transfer it into a shuttle plasmid which can then be used to transform mycobacteria -and thus to express the M. leprae antigen in BCG.
While molecular biology is a vast and ever-expanding discipline, I have tried here to sketch some of the basic principles of molecular biology that I believe have relevance to introducing foreign genes and expressing antigens in BCG. We hope that this will lead to the development of new kinds of multivalent vaccines which will be effective in the prevention of leprosy and tuberculosis, and other infectious diseases as well.
A glance at the future
What is the promise of molecular biological approaches to mycobacteria for the future? Let me consider a few possibilities, or perhaps fantasies:
Development of BCG into a multivaccine vehicle. Our long term hope has been to be able to use BCG as a vehicle for introducing and expressing genes for protective antigens of several pathogens capable of providing one-shot immunization against multiple infectious diseases. The potential advantages of BCG as a multivaccine vehicle are well known: a) It has been used in over a billion people with a very low incidence of serious side effects; b) it is one of only two vaccines that can be given at birth; c) it is generally given as a single-dose vaccine and sensitization to mycobacterial antigens can persist for 5-50 years; d) it is the best adjuvant for human use; and e) it is inexpensive, costing only US$0.055 per vaccine dose. Nevertheless, a great deal more must be understood at a molecular level to be able to achieve a high-level, stable expression of foreign antigens in BCG. With appropriate selectable markers being developed that do not involve antibiotics and reasonably high levels of genetic transformation, it should now be possible to test the feasibility of immunizing with different protective antigens for a number of pathogens expressed in recombinant BCG in experimental systems within the next few years. My assessment is that the limiting factor is unlikely to be the molecular biology but. rather, the immunology, particularly the identification of antigens which are protective.
Genetic strategy to grow M. leprae. The fact that M. leprae grow in human macrophages and Schwann cells in vivo but have not been successfully cultured in vitro, suggests that M. leprae may have accumulated mutations or deletions in a number of important genes required for its survival. Jacobs has formulated a strategy to construct DNA " libraries" containing all of the genes of a rapidly growing mycobacterium, such as M. smegmatis, in our shuttle plasmids (Fig. 11). We would then attempt to transform M. leprae with this library containing all of the genes divided among thousands of plasmids. If one M. leprae cell receives a plasmid containing the genes encoding enzymes which produce the same nutrients that are supplied by their macrophage hosts, then it may be possible for that transformed M. leprae cell to grow in cell-free culture. If that could be achieved, it would be a great deal easier to study the biology, genetics, biochemistry, and immunology of M. leprae.
Fig. 11. Genetic strategy to grow M. leprae.
Clearly, an approach can be employed to genetically engineer slow-growing species of mycobacteria, such as M. tuberculosis and BCG, to grow more rapidly in culture.
Genetic strategy for new drug design. Because M. leprae do not grow in culture and because it is so cumbersome to employ the mouse-foot-pad assay to test whether or not drugs are effective in killing the organism, it is very difficult to design better drugs to be targeted to enzyme targets. Our hope would be to identify potential targets for antibiotics, e.g., topoisomerase as targets of naladixic acid and fluoroquinolines, RNA polymerase as target for rifampin, etc., and then to make mutations in these enzymes in a rapidly growing mycobacterium. The strategy would then be to complement those mutations in the rapidly growing mycobacterium by transforming them with libraries of DNA from M. leprae or M. tuberculosis. Once again, any plasmid which contains the right segment of the M. leprae chromosome that expresses the gene for the target enzyme will complement the defect in the mutant rapid grower, and would now permit it to grow once again in culture. We would, thus, have a rapidly growing mycobacterium that serves as a vicarious host for testing drugs against that target enzyme because it requires that M. leprae enzyme to survive. The bactericidal and bacteriostatic activity of drugs and analogs could be readily tested by measuring their ability to inhibit growth or to kill the rapidly growing recombinant mycobacterial host.
Strategy to identify new protective antigens from any pathogen that kills mice. If high levels of expression of foreign antigen could be achieved in BCG, and if recombinant BCG were really a very effective vaccine and immunogen, it might be possible to immunize mice with a mixture of recombinant BCG cells, only a few of which express the protective antigen of the pathogen to be tested. The strategy would again be to prepare genetic libraries of DNA in shuttle plasmids capable of expressing all of the genes of a pathogen, e.g.. murine malaria or M. tuberculosis, into BCG. The plasmids would be transformed into BCG yielding recombinant BCG. each cell of which expresses a few proteins of the pathogen, but the total library or aggregate represents all of the proteins of the foreign pathogen. These transformed BCG expressing all the antigens of the pathogen would be used to immunize mice, and then the mice would be challenged with a lethal infection of the particular parasite. The only mice that should survive the lethal challenge are those which have been immunized by the recombinant BCG against important protective antigens. The recombinant BCG would be isolated from those survivors, the particular shuttle plasmid containing the gene encoding the protective antigen would be recovered and molecularly characterized. The beauty of this strategy is that the genes for the protective antigen would be cloned even before the nature of the antigen was known.
Rapid new diagnostic tests for leprosy and tuberculosis. It has already been possible to identify unique DNA sequences in M. leprae and in M. tuberculosis that are found only in these species.4-7 Complementary DNA molecules which bind exclusively to these specific characteristic homologous DNA segments are currently being developed using recombinant DNA technology. With such " probes" it is possible to develop tests in which a small DNA probe specific for a unique DNA sequence found in M. leprae, or for a different unique DNA sequence in M. tuberculosis and no other mycobacterium, is used. With a technique called the " polymerase chain reaction" (PCR) one can " amplify," i.e., produce millions of copies of a single DNA sequence in a single cell to a level that can be seen visually or counted by radioactivity labelling techniques (Fig. 12). Thus, if one has a specific DNA probe for M. tuberculosis, it would be possible to bind it to a sample of DNA from a very small number of organisms in a sputum sample, for example, perhaps even one organism. If it found its homolog, it could be amplified several million times and would give a positive signal, indicating that M. tuberculosis was present in the sample. If M. tuberculosis was not present in the sputum, the specific probe would have no complementary DNA sequence to bind, hence, when the PCR amplification was attempted, no signal at all would be produced or amplified. It will thus be possible within a matter of hours to learn if even small numbers of. M. tuberculosis or M. leprae are present within a clinical sample. In the case of tuberculosis, we predict that this will obviate the need for long periods of sputum culture to positively identify the pathogen. It should be possible as well to learn from small biopsies of lesions whether unique DNA sequences found only on M. leprae are present, and to make an earlier and a more certain diagnosis of leprosy. The PCR reaction and other DNA probe amplification techniques are about to revolutionize the diagnosis of many infectious diseases, hopefully including tuberculosis and leprosy. Their principal disadvantage, at present, is primarily the fact that the methodology uses radioisotopes and is currently relatively expensive to set up in the field.
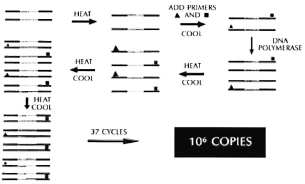
Fig. 12. Polymerase chain reaction (PCR).
This essay has endeavored to share my excitement about the developments in molecular biology, and to outline their power and potential for developing new strategies to combat old infectious diseases. There are many obstacles to be faced, many technical and conceptual problems to be addressed, and many frustrations and failures to be endured. It is only by bringing new tools to bear, however, and asking new questions that, despite inevitable failures, perhaps some new approaches will make meaningful contributions to diagnosing, treating, and ultimately preventing these two ancient scourges of mankind -leprosy and tuberculosis.
- Barry R. Bloom, Ph.D.
Department of Microbiology and Immunology
Albert Einstein College of Medicine
Bronx, New York 10461, U.S.A.
Acknowledgments. Since the writer is not a molecular biologist, whatever merit this article has to contribute reflects the efforts of my many teachers, whom I most gratefully acknowledge. Any errors must indicate that all of the lessons have not yet been learned. It has been a privilege to work with Bill Jacobs, who has essentially created the molecular genetics of mycobacteria and been an inspiring teacher, colleague, and friend. Many of the basic ideas of molecular biology and genetics relevant to mycobacteria were transmitted by Dr. Richard Young at the Whitehead Institute and Dr. Ronald Davis at Stanford University. An inestimable amount of learning came from my students, who tend to question every step and forced me to learn to keep up with them, particularly Scott Snapper, Kalpana Ganjam, Lisa Pane, and Raul Barletta. Special thanks are due to Dr. Laszlo Lugosi, one of the world's grand masters of mycobacteria and BCG, from whom a great deal has been learned. Much of the motivation to think about mycobacteria in molecular terms and most of my understanding about clinical leprosy and tuberculosis has come from my colleagues on the IMMLEP, THELEP, and IMMTUB Steering Committees of the World Health Organization (WHO), and I am particularly indebted to Drs. Noordeen, Grosset. Ivanyi, Engers and Lambert for their limitless help and patience. The support from WHO and the National Institutes of Health that made this work possible is gratefully acknowledged. Finally, I am honored and grateful to have been asked by the organizers of the XIII International Leprosy Congress and to Dr. Anik Rouillon of the IUATLD to contribute this paper.
1. Labidi. A., David. H. L. and Roulland-Dussoix, D. Restriction endonuclease mapping and cloning of Mycobacterium fortuitum var. fortuitum plasmid pAL5000. Ann. Inst. Pasteur Microbiol. 136B (1985) 209-215.
2. Snapper. S. B-, Lugosi. L.. Jekkel, A.. Mellon. R. E.. Kieser. T.. Bloom, B. R. and Jacobs. W. R., Jr. Lysogeny and transformation in mycobacteria: stable expression of foreign genes. Proc. Natl. Acad. Sci. U.S.A. 85 (1988) 6987-6991.
3. Young, R. A. Stress proteins and immunology. Ann. Rev. Immunol, (in press).
4. Clark-Curtiss, J. E. and Docherty, M. A. A species-specific repetitive sequence in Mycobacterium leprae DNA. J. Infect. Dis. 159 (1989) 7-15.
5. Grosskinsky, C. M., Jacobs, W. R., Jr., Clark-Curtiss. J. E. and Bloom, B. R. Genetic relationships between Mycobacterium leprae, Mycobacterium tuberculosis and candidate leprosy vaccine strains by DNA hybridization: identification of an M. leprae-specific repetitive sequence. Infect. Immun. 57(1989) 1535-1541.
6. Eisenach. K. D., Cave. M. D., Bates. J. H. and Crawford, J. T. Amplification of a repetitive DNA sequence specific for Mycobacterium tuberculosis using polymerase chain reaction. J. Infect. Dis. (in press).
7. Hance. A. J., Grandchamp, B., Levy-Frebault, V., Lecossier, D., Rauzier, J., Bocart, D. and Gicquel, B. Detection and identification of mycobacteria by amplication of mycobacterial DNA. Mol. Microbiol. 3(1989) 843-849.
*Hased on State-of-the-Art Lecture presented at the XIII International Leprosy Congress, 13 September 1988, The Hague, The Netherlands. This paper has also been submitted to, and is being published by, the Bulletin of the International Union Against Tuberculosis.