- Volume 57 , Number 2
- Page: 483–91
Reactive oxygen intermediates inactivate Mycobacterium leprae in the phagocytes f rom human peripheral blood
ABSTRACT
Reactive oxygen intermediates such as hydrogen peroxide, superoxide, and hydroxyl radicals are important microbicidal components, and they could also play a role in an infection with Mycobacterium leprae. A comparative study of the level of hydrogen peroxide and superoxide produced by peripheral blood phagocytes f rom normal healthy individuals and lepromatous leprosy patients showed a deficiency in superoxide production in the patients. In the phagocytes f rom normal healthy individuals, there was good release of superoxide ions, and this mediated the killing of M. leprae. The lack of superoxide production allowed the viability of M. leprae inside the macrophages f rom leprosy patients. This deficiency could be rectified by the use of an immunomodulator, the delipidified cell wall of M. leprae. This modulation resulted in the ability of the patients' phagocytes to respond to M. leprae, to produce reactive oxygen intermediates such as superoxide, and also to kill the bacteria. These observations indicate that delipidified cell wall could have significant potential to positively modulate the immune-deficient cells of leprosy patients.RÉSUMÉ
Les intermédiaires oxygénés réactifs, tels que le peroxyde d'hydrogène, le superoxyde, et les radicaux hydroxylés, ont un pouvoir microbicide important; ils pourraient également jouer un rôle dans l'infection par Mycobacterium leprae. Une étude comparative des taux de peroxyde d'hydrogène et de superoxyde produits par les phagocytes du sang périphérique chez des individus normaux en bonne santé et chez des malades atteints de lèpre lépromateusc. a révélé un déficit dans la production de superoxyde chez les malades. Dansles phagocytes provenant d'individus normaux en bonne santé, on a constaté une libération satisfaisante d'ions superoxydes, qui intervenaient dans la lysc de M. leprae Vu l'absence de production de superoxyde, M. leprae peut dès lors survivre à l'intérieur des macrophages des malades de la lèpre. Ce déficit pourrait être corrigé par l'utilisation d'un immuno-modulatcur, à savoir la paroi cellulaire délipidilîée de M. leprae. Cette modulation a conféré aux phagocytes des malades la capacité de répondre à M. leprae, entraînant la production d'intermédiaires oxygénés réactifs, tels que le superoxyde, ce qui entraîne les lyses des bactéries. Ces observations indiquent que la paroi pourrait être dotée du pouvoir de moduler, de manière positive, les cellules immunodéficientes des malades de la lèpre.RESUMEN
Los intermediarios reactivos del oxígeno tales como el peróxido de hidrógeno, el superóxido, y los radicales hidroxilo, son agentes microbicidas importantes y pueden participar en la infección por el Mycobacterium leprae. Un estudio comparativo sobre los niveles de peróxido de hidrógeno y de superóxido producidos por los fagocitos periféricos de individuos normales y de pacientes con lepra, mostró una deficiencia en la producción de superóxido por los pacientes. En los fagocitos de los individuos sanos hubo una buena liberación de iones superóxido y éstos mediaron la muerte del M. leprae. La falta de producción de superóxido permitió la viabilidad del M. leprae dentro de los macrófagos de los pacientes con lepra. Esta deficiencia pudo ser rectificada por el uso de un modulador, la pared celular deslipidizada del M. leprae. Esta modulación capacitó a los fagocitos de los pacientes para responder al M. leprae, para producir intermediarios reactivos del oxígeno tales como superóxido, y también para matar a la bacteria. Estas observaciones indican que la pared celular deslipidizada puede tener el potencial de modular positivamente a las células inmunodeficientcs de los pacientes con lepra.
The microbicidal ability of phagocytes, through reactive oxygen intermediates (ROI), such as hydrogen peroxide (H2O2), superoxide (O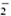 ) and hydroxy! radical (OH·), is a basic defense mechanism of the human host against microbial infection (1,5,11). Some mycobacteria are also identified as susceptible to H2O2 (4,22). There are reports that claim to have demonstrated an inability of the phagocytes from normal healthy individuals to release O on stimulation with Mycobacterium leprae (3), yet others reported that H2O2 and O are produced by the macrophages of leprosy patients in response to agents like phorbol myristatc (PMA) (16). This was thus interpreted that the survival of M. leprae in such patients was due to failure of cell-mediated immunity.
) and hydroxy! radical (OH·), is a basic defense mechanism of the human host against microbial infection (1,5,11). Some mycobacteria are also identified as susceptible to H2O2 (4,22). There are reports that claim to have demonstrated an inability of the phagocytes from normal healthy individuals to release O on stimulation with Mycobacterium leprae (3), yet others reported that H2O2 and O are produced by the macrophages of leprosy patients in response to agents like phorbol myristatc (PMA) (16). This was thus interpreted that the survival of M. leprae in such patients was due to failure of cell-mediated immunity.
It may be recalled that in the absence of a rapid in vitro test to determine the viability of M. leprae, the role of ROI in killing the pathogen inside the host phagocytes could be uncertain. The only report of susceptibility of M. leprae to H2O2 has been by demonstration of the loss of viability of in vitro H2O2 treated M. leprae, by their failure to grow in the mouse foot pad (6,17). Thus, it appears that the reports on the role of ROI in relation to M. leprae inside the phagocytes are incomplete and speculative.
Only a few rapid in vitro tests are available to determine the viability of phagocytosed M. leprae inside macrophages from human peripheral blood. Some of these are adopted and applied in this report. Further, we have earlier established that these in vitro tests are reliable by proving that those samples of M. leprae identified as nonviable by these tests were also found to be nonviable in the classical mouse foot pad tests carried out in parallel experiments (8).
We have determined that H2O2 is induced well in the phagocytes of lepromatous leprosy patients in the presence of live M. leprae but that superoxide ions are not produced (9). However, phagocytes from normal healthy individuals produce both H2O2 and O (10). Thus, if H2O2 should induce killing of M. leprae, then we should sec a loss of viability by our tests in the cells of both normal and leprosy patients. But if both O and H2O2 are needed, then we would sec killing only in the macrophages of the normal healthy individuals and not in those from lepromatous leprosy patients. We were able to test this concept, and this paper reports the results.
If we can modify the macrophages from lepromatous leprosy patients to respond to M. leprae by producing O , then the role of O in relation to M. leprae can be further clarified. This has been possible due to the following reasons: We have been successful in isolating and identifying a component of M. leprae, the delipidificd portion of the cell wall (DCW), as an immunomodulating agent. The potentiality of DCW has been reported in detail (14,21). If the deficiency of superoxide production in leprosy patients is rectified by DCW, then its role in relation to the inhibition or killing of M. leprae could be determined. We report the results of experiments using DCW stimulation.
MATERIALS AND METHODS
Patients. Leprosy patients reporting to the Acworth Leprosy Hospital, Bombay, India, formed part of the study. The patients were classified on the basis of the Ridley and Jopling system (13). The lepromatous group that formed one part of our study was of the long-term-treated (> 4 years), bacillary-negative type (B(-)LL). They were identified as bacteriologically negative when bacilli were undetectable at multiple skin sites and in the nose, and they had had a record of regular treatment in the clinic. These patients were not on treatment at the time of this study.
The normal healthy subjects were volunteers living in the same environment who may have had varied exposure to M. leprae in the city of Bombay. These healthy individuals were neither close contacts nor had regular closeness with leprosy patients.
M. leprae were obtained from tissues, such as the spleen and liver, from infected armadillos obtained from Dr. E. Storrs, Melbourne, Florida, U.S.A. The tissues were collected under aseptic conditions and transported under dry ice to Bombay within a period of 1 week. The tissues were aliquotcd and stored at - 90ºC until needed.
For obtaining host tissue-free M. leprae suspensions, the infected pieces were repeatedly rinsed in sterile normal saline to clute the bacilli. The suspension was centrifuged first at 500 x g x 10 min to remove any larger debris, and later the supernatant was centrifuged at 4500 x g x 30 min. This procedure sedimcntcd the acid-fast stainable bacteria. The bacterial suspension showed no catalase activity and, therefore, was considered to be free from host-tissue contaminants. Catalase is a component of host tissue and is not produced by M. leprae.
Macrophage culture from peripheral blood was prepared as follows. Blood (150 ml) was collected in sterile bottles containing 10 ml of a mixture of 20 units/ml of heparin (Biological Evans, India) and 6% dcxtran (Rallis India, Bombay). The blood was allowed to settle at 37ºC for 45 min. Plasma along with the buffy coat was transferred to a sterile tube and centrifuged at 800 x g x 5 min. The cells were suspended in culture medium after washing them once with minimum essential medium (MEM; Gibco, U.K.). The culture medium consisted of MEM containing human AB-type serum (added at 40% concentration). An aliquot of 5 ml was added to each 55-mm diameter sterile Falcon petri dish. After a 24-hr incubation of the cells at 37ºC in a humidified 5% CO2 atmosphere, the nonadherent cells were removed by draining the liquid. The culture medium was changed thereafter every 48 hr. This resulted in a fairly uniform layer of adherent, esterasepositive phagocytic macrophages after 8 days of culture maintenance. In the above culture conditions we obtained 0.8-1.0 x 106 macrophages per culture dish. In cases where the cells were harvested for an experiment, the cell count was determined before use and the results were expressed as product obtained per 106 macrophages.
H2O2 was estimated following the method of Pick and Mizcl (12) and adopted to mature macrophage cultures. Phenol red solution (Phenol red; Sigma Chemical Co., St. Louis, Missouri, U.S.A.) 0.2 mg/ml and horseradish peroxidase (Sigma) at 2 units/ ml in Eagle's balanced salt solution (EBSS) along with 50 x 106 M. leprae (live or heat killed) were added to mature macrophage culture dishes. These were incubated for 3 hr at 37ºC. The reaction was stopped by the addition of 20 μl of 1 N NaoH, and the color intensity was recorded as the optical density (OD) in a spectrophotometer at 600 nm. The quantity of H2O2 released was expressed as nmol/hr/106 cells using a standard curve for H2O2 and determining the number of cells in each dish after the experiment.
Superoxide was estimated by the method reported by Sugimoto, et al. (19). To well matured macrophages 1 ml of 0.3% nitrobluc tetrazolium (Sigma) was added to each Falcon dish along with 50 x 106 M. leprae (live or heat killed). To one set of cultures, superoxide dismutasc (SOD) (Canine blood; Sigma) at a concentration of 100 μg/ml was added and incubated at 37ºC for 3 hr. The cells were seraped off, counted, and together with cxtracellularly reduced formazan were pelleted at 1000 x g. The pellet was dissolved in 2.5 ml of pyridine and the OD was recorded at 515 nm. The amount of SOD-removable Of was determined by using an extinction coefficient of 2.6 x 104 M-1 cm-1 and expressed as nmol/hr/106 cells.
The scavengers used to remove various oxygen intermediates were 2000 units of catalase (Sigma), 100 μg of SOD (Sigma), 20 mM of sodium benzoate (Ranbaxy Laboratories, India), and 1 mM thiourea (Sisco Research Lab., Bombay). Since sufficient concentrations of these chemicals have been used, it is presumed that effective levels were achieved both inside and outside the phagocytes.
In vitro viability inside phagocytes. M. leprae (25 x 106) were added to well-matured macrophage cultures maintained in sterile Falcon dishes. After 48-hr maintenance of the bacteria inside the phagocytes, their viability was determined by various methods.
One method used to determine the viability of M. leprae inside the macrophages was the fluorescein diacetate (FDA) staining method described earlier (8), which was a modification of that of Kvach and Veras (7). The cells with phagocytosed M. leprae were seraped off the culture dish and lysed with 10 freezing-thawing cycles to release the bacilli. A suspension of the M. leprae (0.001 ml) was placed on a slide and incubated with FDA (4 μg/ml) and ethidium bromide (EB) (4 μg/ml) in the dark for 30 min. The suspension was then examined with a fluorescence microscope under ultraviolet (Fluoval II). A parallel sample from the same preparation was smeared on a slide and stained for acid-fast bacilli (AFB). The number of AFB gave the number of total bacilli and the green fluorescing bacilli in the suspension, as indicated by FDA staining, gave the proportion of bacilli that showed the breakdown of FDA as an indication of viability.
For the labeled uracil incorporation assay, 3H-uracil (Amersham, U.K.; sp. act. 35 Ci/nmol) at a concentration of 5 μCi/dish was added 24 hr after the addition of M. leprae and maintained for 6 days at 37ºC in a humidified 5% CO2 atmosphere. On the sixth day, the medium was removed, the cells were seraped off the petri dishes, and suspended in 1 ml of saline. Cell lysis was carried out by 10 freezing-thawing cycles. M. leprae were released from the macrophage lysate by centrifugation at 4500 x g x 30 min. The bacteria were then washed with 20% trichloroacetic acid to remove the acid-soluble radioactivity. The precipitated material was washed with methanol, suspended in Bray's fluid, and the level of incorporation was determined in a Kontron MR 300 scintillation counter. Heat-killed M. leprae were used as controls. Since there was a wide variation in the incorporation between experiments, the data from individual experiments are presented. Comparison of the results obtained in any one experiment appeared more useful than comparisons of mean values. The method followed here was developed by Vcjare and Mahadevan (20). A similar technique was used by Rook (15) to study the viability of BCG inside phagocytes.
Loss of viability of M. leprae phagocytosed by mature macrophages was also determined by recovering the bacilli after the necessary incubations inside the cells and testing their viability in the foot pads of mice. The recovery of the bacilli was as done after 10 freezing-thawing cycles and centrifugation. The bacterial suspension of 1 x 104 bacilli was used, and the growth obtained at 8 months and 12 months postinoculation was determined. The method of counting the bacillary load in the foot pad was that of Shepard and McRae (18). This test was specifically done to correlate the loss of viability with the production of superoxide ions.
In all experiments where bacteria were obtained by freezing-thawing, the loss of viability due to this technique itself could be determined by comparing the viability of the original bacillary suspension used for phagocytosis.
Preparation of DCW-stimulated culture supernatants. DCW of M. leprae as prepared by the method reported by Robinson and Mahadevan (M) was used from the stock suspension that was available in our laboratory. Briefly, DCW was prepared by sonication (55 watts power) of M. leprae from armadillo tissues for a period of 2-2% hr with pulse sonication of 5 min while the bacteria were kept in an ice bath. The residue obtained after sonication was dclipidified by repeated treatment (3 times) with chloroform methanol (2:1) followed by acetone and either (1:1) at room temperature. The residue was centrifuged, homogenized, and used after determining the level of protein in the suspension. The material was kept sterile during the processing and recovery.
Mononuclear cells were separated over ficoll triosil (Lymphoprep-Nyegaard & Co., Oslo, Norway) density gradient and the cell count was determined. The cell suspension was adjusted to 1 x 106 cells/ml in culture medium (MEM + 20% human AB-type serum) and distributed in pctri dishes. One set of cultures was stimulated with DCW (400 μg as protein for 1 x 106 cells). Incubation was carried out for 96 hr at 37ºC in 5% CO2 atmosphere. The culture supernatant was obtained by filtration (Millex filter; 0.22 μ). The supernatant was stored at -20ºC until further use but used within 4 days to avoid possible loss of stimulatory activity.
Loss of M. leprae viability in macrophage cultures exposed to DCW-stimulated culture filtrate. To the mature macrophage culture, the full 1 ml of DCW-stimulated culture supernatant (from the same patient) was added and incubated for 24 hr at 37ºC. At the end of the incubation period, the medium was aspirated and fresh medium along with M. leprae (25 x 106) was added. At that time, different treatments (addition of catalasc, SOD, benzoate, etc.) were carried out, and phagocytosis was allowed for 48 hr at 37ºC in a humidified 5% CO2 atmosphere. The cultures were terminated, the bacilli were released by freezing-thawing 10 times as described earlier, and the viability determined by the methods mentioned before. In a parallel culture, the ROI released were also determined with special reference to superoxide.
Results are expressed as average values with standard deviations (± S.D.). Statistical significance was determined by Student's t test.
RESULTS
Mature macrophages from the peripheral blood of normal healthy individuals are able to produce notable levels of FLO, (111.2 ± 10.5 nmol/hr/106 cells) and O (2.2 ± 0.6 nmol/hr/106 cells) when challenged with live M. leprae. On the other hand, macrophages from long-term-treated, bacteriologically negative patients (B( -)LL) responded to M. leprae by producing H2O2 (89 ± 2.6 nmol/ hr/106 cells), but produced very low levels of superoxide (0.3 ± 0.2 nmol/hr/106 cells). The lower threshold level of sensitivity of superoxide estimation in our assay was 0.4 nmol/106 macrophages. The superoxide level in Table 1 is expressed as SOD-removable O, and the levels obtained in eight separate experiments have been expressed as mean values by subtracting the level of Of with SOD from that obtained without SOD. The phagocytic activity of the macrophages from the patients was much higher than that of the cells from normal individuals (Table 1).

Table 2 shows that along with phagocytosis of the bacteria by the cells there was a loss of viability as measured by FDA-EB staining. It had been observed that there was a significant loss of viability as measured by FDA-EB staining of the bacteria inside the macrophages by just freezing and thawing, as is seen in Table 5. In this experiment (Table 2), phagocytosis was associated with H2O2 and O production and, specifically, the level of O released averaged 1.8 nmol/ hr/106 cells. There was a reduced loss of viability after phagocytosis when either SOD or catalase, or both of the scavengers of ROI, were added. Since both catalase and SOD maintained good viability as measured by FDA-EB staining, it was appropriate to determine the role of OH· radicals by using an OH· antagonist such as sodium benzoate. The addition of sodium benzoate appeared to block the killing of M. leprae in all three experiments in which it was tried, suggesting a probable role for OH·.
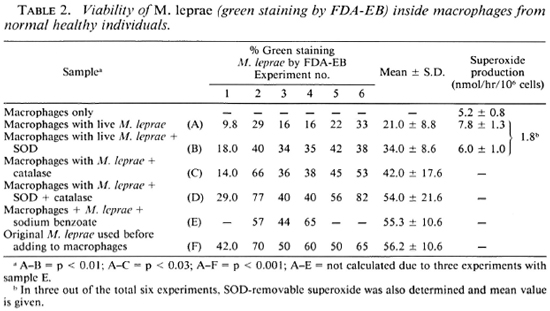
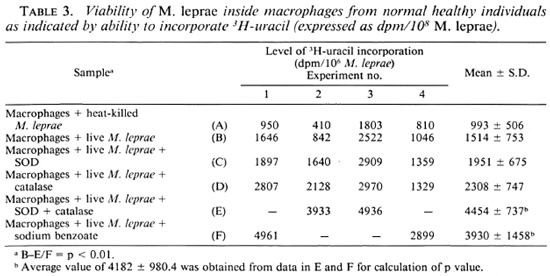
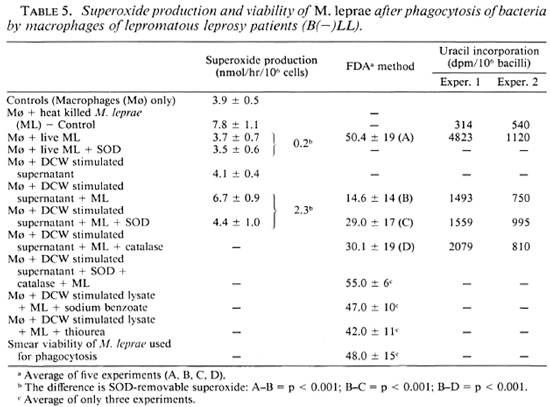
We also examined the ability of M. leprae to take up labeled uracil as an indicator of viability (20). Table 3 presents the data which indicate the level of incorporation of labeled uracil by M. leprae after phagocytosis. This level is not statistically different from that of the uptake of uracil by heat-killed M. leprae phagocytosed by the macrophages. However, if one compares the level of in corporation in the four separate experiments, in two of them SOD and catalase were used and in another two sodium benzoate was used, the level of incorporation appears to be higher compared to the control (with live M. leprae).
Lastly, we confirmed the loss of M. leprae viability inside the macrophages of healthy controls by recovering the phagocytosed bacteria 48 hr after phagocytosis and injecting 1 x 104 of these bacilli into the foot pads of at least 15 Swiss white mice. This was compared with the M. leprae obtained under various conditions of treatment of the macrophages. Data presented in Table 4 show that the M. leprae obtained after incubation inside the macrophages showed no multiplication. The minimum number of bacilli/foot pad that could be detected was 7.5 x 103 by our method; thus the 1 x 104 inoculum had not multiplied further. Those bacteria phagocytosed by macrophages and maintained in the presence of SOD or catalase, however, did show growth at the 8 and 12-month harvests, indicating survival of the phagocytosed M. leprae.
Since a low O level is demonstrable in the macrophages from leprosy patients, one could expect survival of M. leprae inside these cells. The data presented in Table 5 show that with a low level of SOD-removable O (0.2 nmol/hr/106 cells), the viability of M. leprae after phagocytosis was as good as the viability of bacilli used for the experiment. This was in spite of a good production of H2O2 as indicated by the data presented in Table 1. It thus appears that in spite of the ability to produce H2O2, the viability of M. leprae is maintained. This could be due to a low production of O.
When the culture filtrate from DCW-stimulated leukocyte cultures from patients was added to the patients' own macrophages, the cells responded well to M. leprae and produced SOD-removable O (Table 5). Along with this, the viability of M. leprae was reduced from 50% to 14%, as measured by the FDA-EB test system. This correlated with a lowered incorporation of labeled uracil by M. leprae (Table 5). Such a loss of viability as measured by FDA-EB could be prevented by SOD and catalase as well as sodium benzoate or thiourea (Table 5).
DISCUSSION
The virulence of leprosy bacilli and the inherent microbicidal ability of the macrophages that ingest them are important parameters in establishing leprosy infection. Immune processes are also called into play soon afterward. A rapid accumulation and activation of macrophages occur in hosts possessing cell-mediated immunity. The rapid accumulation of the immune-activated macrophages usually inhibits the growth of the bacilli, thereby preventing clinical disease. If these macrophages do not acquire enough microbicidal abilities, the bacilli will grow intracellulary and soon the disease will express itself. This is what is observed in lepromatous leprosy.
The results in these studies indicate that given the same ratio of M. leprae to the cells, the macrophages from the normal individuals were stimulated by live M. leprae to produce O and H2O2; whereas the cells of B(-)LL patients have a very low superoxide stimulatory ability. Thus, in normal healthy individuals live M. leprae encountering host cells may be killed by ROI produced by macrophages when the bacteria are being phagocytosed. In a recent paper, Holzcr, et al. (2) showed that M. leprae are able to induce a low but significant level of superoxide in the macrophages of normal healthy individuals and also phagocytose the bacteria slowly. This was a modification oftheir earlier report (3). Cell-mediated immunity may also play a role as a subsequent event. In the minority who express lepromatous leprosy, the bacteria survive after phagocytosis by the macrophages, perhaps due to poor O and OH· radicals. Thus, both cell-mediated immunity and ROI defects exist. This observation has been confirmed by the data presented here.
The survival and killing of M. leprae inside the phagocytes have been determined by two in vitro tests as well as in the mouse foot pad. Without relying on only any one test, if we generally compare all of the three test systems it appears that the observations of survival or killing have given us a true picture in relation to reactive oxygen intermediates. It appears most likely that the cells exposed to DCW are able to be activated through cell cooperation, and the activated macrophages are then able to kill the M. leprae through reactive oxygen species.
The importance of ROI and the deficiency in these components in lepromatous leprosy patients were further confirmed. When such deficient cells were exposed to culture supernatants from DCW-stimulated leukocyte cultures from the same patient, the cells attained an ability to produce O while recognizing M. leprae and then to kill the bacteria.
Earlier, we have reported the immunomodulatory ability of DCW in mice and human cells tested in vitro (14,21). In recent experiments (unpublished), we have shown that mice injected with DCW showed activated macrophages in the peritoneal cavity. Such macrophages were able to inactivate M. leprae in an in vitro culture, something not seen with the macrophages of nonimmunized mice. It appears clearer now how our earlier observation of the killing of M. leprae inside the macrophages of leprosy patients could have been mediated.
By relating all these observations, it is possible to predict that DCW is recognized as an antigen by the immune-deficient cells of leprosy patients, and the cells can then develop the ability to induce cell-mediated immune reactions leading to the production of active lymphokincs which could, in turn, activate the deficient macrophages. Such activated macrophages are able to handle M. leprae as the macrophages from normal healthy individuals do through reactive oxygen intermediates. Several recent observations (unpublished) also support this prediction.
Such an event and process, if well documented, could then add immense potentiality to DCW as a powerful immunomodulator with the possibility for use in leprosy patients.
Acknowledgments. The authors wish to express their gratitude to Dr. E. Storrs, Melbourne, Florida, U.S.A., for the supply of infected armadillo tissues (aided by LEPRA, U.K.) and to Acworth Leprosy Hospital, Wadala, Bombay. India, for providing us with human materials. The work was carried out under grant no. D71/85 approved and financed by the Department of Science and Technology. Government of India.
REFERENCES
1. BABIOR, B. M. Oxygen-dependent microbial killing by phagocytes. N. Engl. J. Med. 298(1978)659-668.
2. HOLZER, T. J., KIZLAITIS, L., VACHULA, M., WEAVER, C. W. and ANDERSEN, B. R. Human phagocytic cell responses to Mycobacterium leprae and Mycobacterium bovis bacillus Calmette-Guérin; an in vitro comparison of leprosy vaccine components. J. Immunol. 141(1988)1701-1708.
3. HOLZER, T. J., NELSON, K. E., SCHAUF, E., CRISPEN, R. G. and ANDERSEN, B. R. Mycobacterium leprae fails to stimulate phagocytic cell superoxide anion generation. Infect. Immun. 51(1986)514-526.
4. JACKETT, P. S., AHER. V. R. and LOWRIE, D. B. Virulence and resistance to superoxide, low pH and hydrogen peroxide among strains of Mycobacterium tuberculosis. J. Gen. Microbiol. 104(1978)37-45.
5. KLEBANOFF, S. J. Oxygen-dependent cytotoxic mechanisms of phagocytes. Adv. Host Def. Mech. 1(1982)111-162.
6. KLEBANOFF, S. J. and SHEPARD, C. C. Toxic effect of the peroxidase-hydrogen pcroxidc-halide antimicrobial system on Mycobacterium leprae. Infect. Immun. 44(1984)534-536.
7. KVACH, J. T. and VERAS. J. R. A fluorescent staining procedure for determining the viability of mycobacterial cells. Int. J. Lepr. 50(1982)183-192.
8. MAHADEVAN, P. R., JAGANNATHAN, R., BHAGRIA, A., VEJARE, S. and AGARWAL, S. Host-pathogen interaction -new in vitro drug test systems against Mycobacterium leprae-possibilities and limitations. Lepr. Rev. 57 Suppl. 3(1986)182-200.
9. MAROLIA, J. and MAHADEVAN, P. R. Superoxide production from macrophages of leprosy patients after stimulation with Mycobacterium leprae. J. Biosci. 12(1987)273-279.
10. MAROLIA, J. and MAHADEVAN, P. R. Mycobacterium leprae mediated stimulation of macrophages from leprosy patients and hydrogen peroxide production. J. Biosci. 13(1988)295-303.
11. NATHAN, C. F., NOGUEIRA, N., JUANGBHANICH, C, ELLIS, J. and COHN, Z. A. Activation of macrophages in vivo and in vitro. Correlation between hydrogen peroxide release and killing of Trypanosoma cruzi. J. Exp. Med. 149(1979)1056-1068.
12. PICK, E. and MIZEL, D. Rapid microassays for the measurement of superoxide and hydrogen peroxide production by macrophages in cultures using an automatic enzyme immunoassay reader. J. Immunol. Methods 46(1981)211-226.
13. RIDLEY, D. S. and JOPLING, W. H. Classification of leprosy according to immunity; a five-group system. Int. J. Lepr. 34(1966)255-273.
14. ROBINSON, P. and MAHADEVAN, P. R. A component of Mycobacterium leprae as immunomodulating agent for immune deficient cells of leprosy patients. J. Clin. Lab. Immunol. 24(1987)171-176.
15. ROOK, G. A. W. An isotope incorporation assay for the antibacterial effects of human monocytes. Ann. Immunol. 132D(1981)281-289.
16. SHARP, A. K. and BANERJEE, D. K. Hydrogen peroxide and superoxide production by peripheral blood monocytes in leprosy. Clin. Exp. Immunol. 60(1985)203-206.
17. SHARP, A. K., COLSTON, M. J. and BANERJEE, D. K. Susceptibility of Mycobacterium leprae to the bactericidal activity of mouse peritoneal macrophages and to hydrogen peroxide. J. Med. Microbiol. 19(1985)77-84.
18. SHEPARD. C. C. and MCRAE. D. H. A method for counting acid-fast bacteria. Int. J. Lepr. 36(1968)78-82.
19. SUGIMOTO, M., HIGUCHI, S., ANDO, M., HORIO, S. and TAKUOMI. H. The effect of cytochalasin B on the superoxide production by alveolar macrophages obtained from normal rabbit lungs. J. Reticuloendothel. Soc. 31(1982)117-130.
20. VEJARE, S. and MAHADEVAN, P. R. Importance of determining viability of Mycobacterium leprae inside macrophages -an in vitro method using uracil. J. Biosci. 11(1987)455-463.
21. VERMANI, M. and MAHADEVAN, P. R. A component of Mycobacterium leprae as immunomodulaling agent -activation of macrophages to contain viability of M. leprae in vivo and in vitro. Health Coop. Papers 7(1987)153-161.
22. WALKER, L. and LOWRIE. D. B. Killing of Mycobacterium microti by immunologically activated macrophages. Nature (Lond.) 293(1981)69-70.
Ph.D., The Foundation for Medical Research, 84-A R.G. Thadani Marg, Worli, Bombay 400018, India.
Reprint requests to Dr. Mahadevan.
Received for publication on 20 April 1988.
Accepted for publication in revised form on 20 January 1989.