- Volume 68 , Number 1
- Page: 27–39
Antileprosy protective vaccination of rhesus monkeys with BCG or BCG plus heat-killed Mycobacterium leprae: immunologic observations
ABSTRACT
Groups of rhesus monkeys were vaccinated and boosted with Mycobacterium bovis bacillus Calmcttc Gucrin (BCG) or BCG plus low-dose (LD) or high-dose (HD) heat-killed M. leprae (HKML), or were unvaccinated. Prior to and following vaccination-boosting and subsequent M. leprae (ML) challenge, these and unvaccinated, unchallenged control monkeys were observed longitudinally for approximately 3 years. Vaccination with BCG plus HKML initially stimulated significant in vitro blood mononuclear cell blastogenic responses to lepromin, which returned to baseline postboosting and post-Iive-ML-challenge, minimally reappearing significantly 2 years post-ML-challenge. Vaccination with BCG failed to stimulated positive blastogenic responses to lepromin before ML-challenge but small, marginally positive, intermittent responses were seen post-ML-challcnge. Compared to the unvaccinated ML-challenged group, significant increases in the numbers of blood CD4+ and CD8+ T-ccll subsets and an increased CD4+:CD8+ ratio were observed in both BCG plus HKML-vaccinated, ML-challenged groups, bu t no t in the BCG-onlγ-vaccinatcd, ML-challenged group. CD4+CD29+ and CD4+CD45RA+ subset numbers increased significantly over time in only the BCG lenged groups, vaccination with BCG or BCG plus HKML followcd by ML-challcnge produced lower lgM:IgG antiphenolic glycolipid-I (PGL-I) serum antibody ratios and protected rhesus monkeys í'rom clinicai leprosy, consistent with prior observations that low IgM:IgG anti-PGL-I responses correlated with resistance to and protection f rom leprosy.RÉSUMÉ
Des groupes de macaques rhesus furent vaccinés par Mycobacterium bovis, bacille de Calmette et Guérin (BCG) de ou BCG accompagné d'une faible dose (FD) ou d'une haute dose (HD) de M. leprae inactivées par la chaleur (MLIC), ou ne furent pas vaccinés. Ces animaux furent ensuite inoculés par M. leprae (ML), excepté un groupe contrôle de singes nonvaccinés et non inoculés. Tous furent suivis longitudinalement pendant approximativement 3 années. Rapidement après la première injection et l'injection de rappel, le vaccin associant BCG et MLIC a stimulé, de façon significative, une prolifération des cellules mononuclées in vitro en réponse à la lépromine, qui est rapidement retournée à des valeurs basales après injection de rappel et après inoculation avec des ML virulentes. Cette réponse proliferative réapparut, de façon minimale mais significative, 2 ans après l'inoculation de ML. La vaccination avec le BCG n'a pas permis d'obtenir une réponse proliferative blastogénique à la lépromine avant l'inoculation avec ML mais une réponse limitée, intermittente positive de façon marginale fut observée après inoculation de ML. En comparaison du groupe inoculé mais non vacciné, les groupes vaccinés avec BCG associé aux MLIC, les groupes inoculés avec ML, mais pas le groupe vacciné avec BCG seul et inoculé, ont montré une augmentation significative du nombre des cellules T circulantes de type CD4+ et CD8+ et du ratio CD4+:CD8+. Les sous-populations CD4+CD29+ et CD4+CD45RA+ augmentèrent signilicativement au cour du temps seulement chez le groupe vacciné avec BCG plus MLIC FD et inoculé avec ML. En comparaison du groupe non-vacciné et inoculé avec ML, la vaccination avec le BCG ou le BCG plus MLIC précédent l'inoculation avec ML a été à l'origine d'un rapport plus bas du ratio IgM:IgG des anticorps sériques dirigés contre les glycolipides phénoliques de type I (PGL-1) et a protégé les macaques rhésus contre le développement de lèpre clinique. Ceci est en accord avec les observations précédentes indiquant qu'une réponse caractérisée par un rapport d'IgM:IgG bas d'anticorps anti-PGL-I corrèle avec résistance et protection contre la lèpre.RESUMEN
Se vacunaron monos rhesus con Mycobacieriiun bovis BCG y se retaron con BCG, o con BCG más dosis bajas (DB) de M. leprae muerto por calor (MLMC), o con BCG más dosis altas (DA) de MLMC. Algunos monos se mantuvieron sin vacunar. Los monos de todos los grupos experimentales se mantuvieron bajo vigilancia durante 3 años, aproximadamente. La vacunación con BCG más MLMC inicialmente estimuló una significante respuesta linfoproliferativa contra la lepromina, la cual regresó al valor basal encontrado después de la reestimulación y el reto con MLM vivo, reapareciendo en forma mínima 2 años después del reto con ML. La vacunación con BCG no estimuló la respuesta blastogénica a la lepromina antes del reto con ML pero se observaron pequeñas respuestas marginalmente positivas e intermitentes después del reto con ML. Comparado con el grupo no vacunado retado con ML, se observó un incremento significativo en los números de células T CD4+ y CD8+ y una relación CD4+:CD8+ aumentada tanto en el grupo vacunado con BCG más MLMC como en el retado con ML, pero esto no se observó en el grupo vacunado sólo con BCG y retado con ML. La cantidad de células CD4+CD29+ y CD4+CD45RA+ aumentó significativamente con el tiempo sólo en el grupo vacunado con BCG más DAML retado con ML. Comparando con el grupo no vacunado retado con ML, la vacunación con BCG o con BCG más MLMC, seguida por el reto con ML, produjo relaciones IgM:IgG más bajas de anticuerpos antiglicolípido fenólico-I (PGL-I) y protegió a los monos rhesus de la lepra clínica: hallazgos que fueron consistentes con observaciones previas que indicaron que las bajas respuestas IgM:IgG anti-PGL-I, correlacionaron con la resistencia y protección contra la lepra.It is well established that vaccination of humans with BCG offers protection from clinical leprosy (7-10,11). The degree of protection has varied, however, from near zero to approximately 80% in human studies in different parts of the world (1,3,5,7, 9,28,30). The possible beneficial effects of combining BCG with heat-killed Mycobacterium leprae (HKML) as a protective vaccine are more controversial (5,10) Our results of vaccination of rhesus monkeys (RM) and sooty mangabey monkeys (SMM) with BCG or BCG + HKML followed by experimental challenge with live M. leprae (ML) suggested that BCG offered protection from clinical leprosy in both species; the addition of HKML to the BCG vaccine produced an additive protective effect in RM over that produced by BCG alone, but produced a greater susceptibility to clinical leprosy in SMM (12).
We have reported that RM, as a group, are prone to paucibacillary (PB) forms of leprosy in at least 75% of cases (18); whereas SMM are prone to multibacillary (MB) forms in at least 80% of cases (15,17,19). The basis of these differences in leprosy susceptibility is not known, nor is it known exactly why some human individuals and human populations are prone to one form of leprosy versus the other.
Taken together, our prior observations suggest that BCG offers some protection from clinical disease at both the PB and MB ends of the leprosy spectrum; the combination of HKML with BCG, however, renders MB-prone SMM more susceptible to leprosy while enhancing the BCG protective effect in PB-prone RM (12). These results suggest that comparative immunologic studies of ML-infcctcd RM versus SMM might offer an avenue to study basic immunologic mechanisms determining resistance/susceptibility characteristics resulting in natural resistance versus partial resistance (PB-prone) versus susceptibility (MB-prone) to leprosy.
RM and SMM are phylogenetically very close to humans, and such studies in monkeys should provide information pertinent to humans. Comparative studies in RM versus SMM before and after vaccination with BCG alone compared to vaccination with BCG + HKML or unvaccinated controls followed by challenge with live ML might also be expected to show important relationships between immunologic parameters that could further help to explain the basis for resistance/susceptibility characteristics of human individuals and populations and the variations observed in the degree of protective efficacy of BCG in different human populations. The data might also reveal unrecognized aspects of the immune response to ML antigens to which future antileprosy vaccines can be targeted for more effective protection.
We report herein the results of longitudinal immunologic observations spanning approximately 3 years in groups of RM before any experimental manipulation, after vaccination with BCG, BCG + low dose (LD) HKML, BCG + high dose (HD) HKML or no vaccination, after boosting with the same vaccines and after experimental challenge of all groups with live ML. The clinical results of these studies were previously reported (l2).
MATERIALS AND METHODS
Animals.
The methods and procedures have been previously described (12,15,17-19). Briefly, 45 Chinese RM (Macaca mulatto), 2-3 years old, born and maintained in our breeding colony at the Tulanc Regional Primate Research Center (TRPRC), Covington, Louisiana, U.S.A., all with presumed similar natural exposures to environmental agents, were divided into 4 experimental groups (3 vaccine groups and 1 unvaccinated control group-all ML-challenged) of 10 (3 females and 7 males per group). There was also one group of 5 unvaccinated, non-ML-challengcd normal controls.
Preparation of HKML for vaccination and live ML for monkey challenge. Details of the preparations were previously published ('-). Briefly, SMM-origin ML was isolated from an SMM and inoculated into armadillos by Drs. Wayne M. Meyers and Gerald P. Walsh at the Armed Forces Institute of Pathology, Washington, D.C., U.S.A. The livers and spleens were taken from these armadillos when leprosy became sufficiently advanced and were stored frozen (-70ºC) until shipment to the laboratory of Dr. Patrick J. Brennan (Department of Microbiology, School of Veterinary Medicine, Colorado State University, Fort Collins, Colorado, U.S.A.) for isolation and purification of ML by the Draper method (6). The ML preparations were heat-killed (autoclaved), lyophilized and shipped to the TRPRC. These procedures were performed in Dr. Brcnnan's laboratory under contract AI-52582 from the National Institute of Allergy and Infections Diseases, National Institutes of Health, Bethesda, Maryland, U.S.A.
Immunizations with BCG or BCG + HKML. Monkeys were vaccinated at the TRPRC with BCG alone or BCG + HD HKML or BCG + LD HKML by intracutaneous (i.c.) injection of 0.1 ml of the appropriate suspension (12). Primary vaccinations were followed by boosting at 5 weeks, and challenge with live ML was at 10 weeks. The three vaccine groups received the following vaccinations and boosters, respectively: 1) BCG alone [1-2.6 x 106 viable units (vu)]; 2) BCG (1-2.6 x 106 vu) + LD HKML (1.6 x 109); and 3) BCG (1-2.6 x 106 vu) + HD HKML (3.2 x 109) (l2).
Monkey inoculations. Monkeys were inoculated with a total of 4.3 x 109 live, freshlγ-prepared ML suspensions by combined i.e. and intravenous (i.v.) routes. The i.e. sites receiving a total of 1.7 x 109 ML equally distributed were: 2 i.e. sites per car, the tip of the nose, outer forearms and outer calves. The i.v. inoculations (2.6 x 109 ML) were made via the saphenous vein. ML suspensions for RM challenge had a morphological index (Ml) of 8%.
ELISA. The ELISAs were performed as previously reported (13,15,19). Natural ML phenoloic glycolipid-I (PGL-I) was used as antigen (Ag). PGL-I was provided by Dr. Patrick J. Brennan under NIH contract AI52582. Data are presented as the group means of longitudinal time points; error bars were omitted to avoid congestion.
Blastogenesis. This procedure for his togenesis has been described in detail previously (13,18). Briefly, heparinized blood was used to prepare buffy coats which were centrifuged on Ficoll/Hypaque, washed and suspended in RPMI-1640 containing 20% heat-inactivated human AB serum, glutamine and penicillin/streptomycin. The mononuclear cell (MNC) fraction was used at 2 x 106/ml for in vitro blastogenesis studies with or without 100 µg/ml of lepromin in U-bottom, 96-well microtiter plates. MNC (2 x 105 per well) were incubated at 37ºC in 5% CO2 in triplicate for 5 days with stimulant or media prior to pulsing for 18 hr with 1 µci of' 3H-thymidine per well. Thereafter, cells were washed and harvested on a cell harvester and quantified by scintillation counting. Stimulants were lepromin (human/armadillo, prepared by Dr. Wayne M. Meyers); Rees soluble ML antigen (gift from Dr. R. J. W. Rees, Medical Research Council, Middlesex, U.K.) and ML rlOkDa protein (provided by Dr. Patrick J. Brennan under NIH contract AI-52582). Results are presented as the group means of stimulation indices (SI) calculated by dividing the disintegrations per minute (dpm) in averaged triplicates in experimental tubes by the average triplicate dpm in the control (unstimulated) tubes. Data arc presented as the group means of longitudinal points (bars representing the standard errors are added to points of statistical importance; error bars were not included for other points to make the graphs easier to read).
Peripheral blood lymphocyte (PBL) subsets. Whole EDTA blood was obtained longitudinally, stained with mouse antihuinan monoclonal antibodies, and examined by flow cytometry, as previously reported p , 14,23.24) Monoclonal antibodies with the following specificities were used: CD4+, CD8+, CD4+CD29+ and CD4+CD45RA+. Results are presented as the group mean number of cell subset/mm1 at different time points. Error bars were not added to these figures due to the problem of congestion, but the statistical data are noted where pertinent in the text.
Statistical analyses. All statistical calculations were performed using statistical programs for the Macintosh Computer. Longitudinal comparisons between groups were performed by MANCOVA analysis. The paired t test was additionally used at selected time points for PBL subset data analysis and for the analysis of blastogenic data.
RESULTS
Figure 1 shows the results of in vitro blastogenic studies spanning a period of approximately 3 years prior to vaccination/boosting and subsequent to live ML challenge. The first two time points are prevaccination baseline points. Significant (p <0.001) dose-dependent responses were noted within 49 days postvaccination in both the BCG + HD HKML and the BCG + LD HKML groups; no blastogenic responses to lepromin were seen in the BCGonly vaccinated or the unvaccinated groups at this time (Fig. 1). Diminished responses to ML antigens were seen in both the LD and the HD HKML groups 20 days postboosting (on the day of ML inoculation, time zero) (Fig. 1). Two months postinoculation (PI), there was a small, insignificant peak (shoulder) response in the BCG + LD HKML group. Four months PI, small insignificant responses to lepromin were observed in the BCG-only and the BCG + LD HKML groups; thereafter, responses in all groups essentially returned to baseline until 24 months PI (Fig. 1). MANCOVA statistical analysis revealed that the blastogenic responses in all three ML-challengcd vaccine groups differed significantly from the unvaccinated, ML-challengcd control group over the course of this study (BCG p <0.02; BCG + LD or HD HKML, p <0.0001). The responses in the vaccinated groups between 5 months and 20 months PI were intermittently marginally positive (Sis ranged from approximately 1.5 to <4 and p values ranged from <0.05-<0.002). At 24 months PI, small significant peak responses were seen in the BCG + LD HKML group (p <0.05) and in the BCG + HD HKML group (p <0.007). Similar patterns were seen in response to Rees soluble ML antigen and ML 10-kDa protein, but they were lower in magnitude than the responses to lepromin (data not shown). During the 32-month PI period covered by these longitudinal studies, as previously detailed, the following numbers of RM out of 10 per group showed signs of clinical leprosy; 9 or 10 unvaccinatcd, depending on definitions; 3 BCG-only; 2 BCG + LD HKML; and 1 BCG + HD HKML (12).
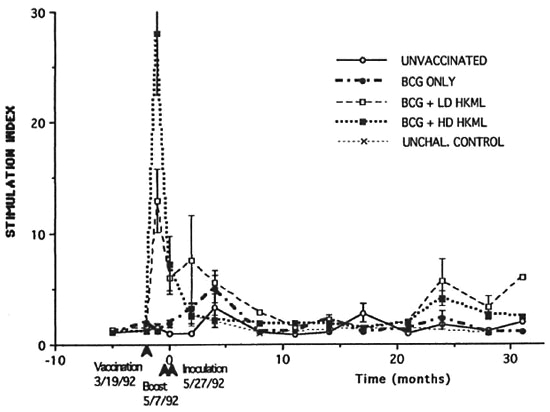
Fig. 1. Longitudinal in vitro blastogenic responses (stimulation indices, SI) of peripheral blood mononuclear cells to lepromin. Each point represents the mean value per group at the indicated time. Standard error bars were omitted from many points for clarity. The first two sets of time points were prevaccinalion (vaccination was done on the day of the second set of time points). The third set of time points are on the day of boosting (49 days postvaccination). The fourth set of time points are on the day of ML-challenge (20 days post-boosting, designated time 0).
Longitudinal monitoring of blood lymphocyte subsets revealed significant sustained increases in the absolute numbers of several T-cell subsets in the blood of all three vaccinated, ML-challenged groups relative to the unvaccinated, ML-challenged group (Figs. 2-5). The p values below refer to the changes in the numbers versus time in vaccinated groups relative to that of the unvaccinated, ML-challenged group. Significant sustained increases were seen in numbers of the CD4+ (helper cell) subset in both the BCG + LD HKML-vaccinated (p <0.02) and the BCG + HD HKML-vaccinatcd (p <0.02) after MLchallcngc; the increase seen in this subset in the BCG-only vaccinated group was not significant (Fig. 2).
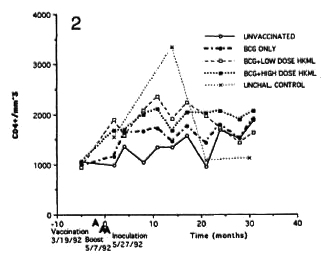
Fig. 2. Longitudinal absolute number (cens per inm3 of blood) of CD4+ cens (mean values per group).
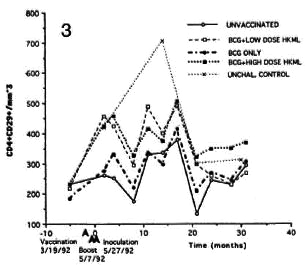
Fig. 3. Longitudinal CD4+CD29+ numbers (see Fig. 2 legend).
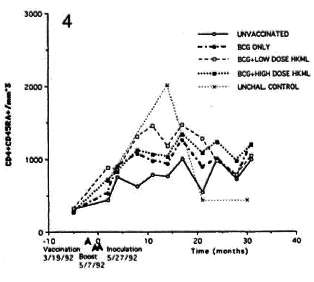
Fig. 4. Longitudinal CD4+CD45+ numbers (see Fig. 2 legend).
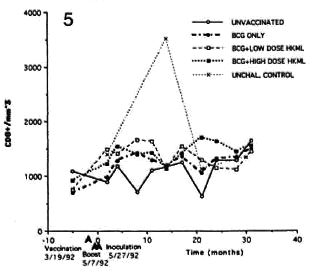
Fig. 5. Longitudinal CDS+ numbers (see Fig. 2 legend).
Significant sustained increase with fluctuations over time were observed in the CD4+CD29+ (helper-inducer) and the CD4+CD45RA+ (suppressor-inducer) subsets in the BCG + LD HKML vaccine "roup (p <0.008), but not in the BCG + HD HKML or the BCG-only groups (Figs. 3 and 4).
Significant differences (p <0.02) were seen, again with fluctuations, in the numbers of CD8+ cells (all but a small percentage of CD8+ cells were CD 16-ncgativc by double staining) in both of the BCG + HKML-vaccinated, ML-challenged groups, but there was an insignificant increase in the BCG-only vaccinated, ML-challenged group (Fig. 5).
Over the period of 8-21 months after ML challenge the CD4: CD8 ratio was significantly increased longitudinally in the BCG + LD HKML (p <0.003) and the BCG + HD HKML groups (p <0.007), but not in the BCG-only vaccinated group, compared to the unvaccinated, ML-challenged group (Fig. 6).
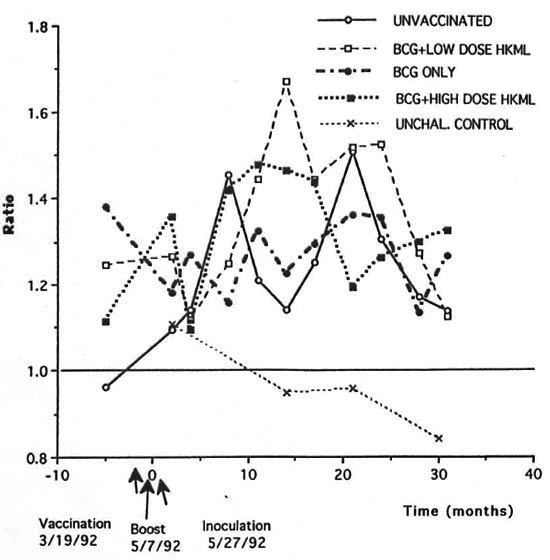
Fig. 6. Longitudinal CD4:CD8 ratios.
Longitudinal ELISA determinations of IgG and IgM antibody to ML-specific PGL-I antigen showed the following:
a) rapid IgG anti-PGL-I responses to vaccination/boosting with BCG + LD or HD HKML which platcaued immediately after ML challenge (time zero) and differed significantly (MANCOVA) from the unvaccinated, ML-challenged group. Beginning 6 months after ML inoculation, the level of IgG anti-PGL-I began a significant decline in the LD and HD"HKML + BCG groups. In the LD HKML group, IgG anti-PGL-I continued to decline to a level below the unvaccinated, ML-challenged control group; IgG anti-PGL-I responses in the BCG-only vaccinated, ML-challenged vs. the unvaccinated ML-challenged group did not differ significantly, appeared only after ML challenge (as expected) and platcaued 6 months PI (Fig. 7).
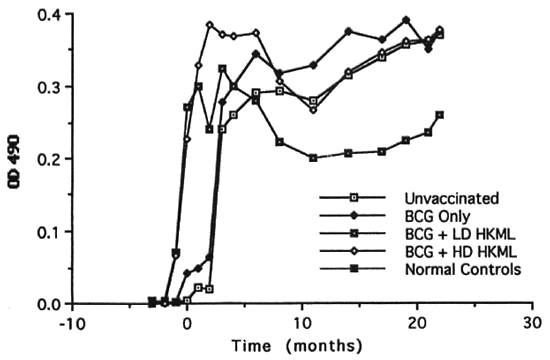
Fig. 7. Longitudinal IgG anti-PGL-I serum antibody levels (mean levels per group).
b) IgM anti-PGL-I (Fig. 8) also rose rapidly after vaccination/boosting with BCG + HKML, platcaued at optical density (OD) levels lower than IgG anti-PGL-I immediately after ML challenge, and then declined rapidly to levels much less than those of the ML-challenged, unvaccinated group; in the BCG-onlγ-vaccinated group, IgM anti-PGL-I rose steeply after ML-challengc, platcaued within 2 months, and then fell steeply; by 4 months PI, IgM anti-PGL-I OD levels also began a rapid decline in the ML-challenged unvaccinated group, eventually (by 6 months PI) plateauing higher than any ML-challcnged vaccinated group. Similar to the IgG anti-PGL-I data, IgM responses in the ML-challenged unvaccinated controls and the BCG-only vaccinated group differed significantly from the BCG + HKML groups and appeared after ML-challcnge (Fig. 8). After ML-challcnge, the ratio of ELISA-dcrivcd OD values for IgM : IgG anti-PGL-I was highest in the unvaccinated, ML-challenged control group > the BCG group > BCG + LD & HD HKML groups, following the pattern of leprosy susceptibility in these groups (Fig. 9)(12).
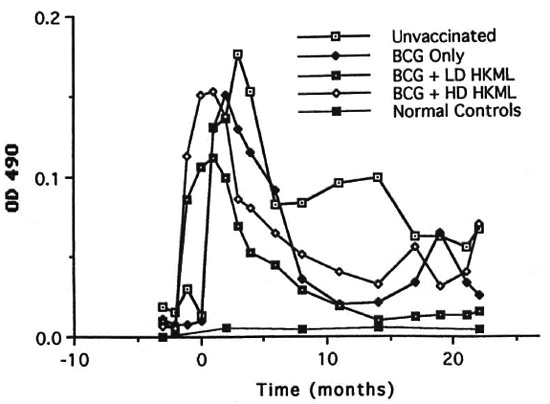
Fig. 8. Longitudinal IgM anti-PGL-I serum antibody levels (mean levels per group).
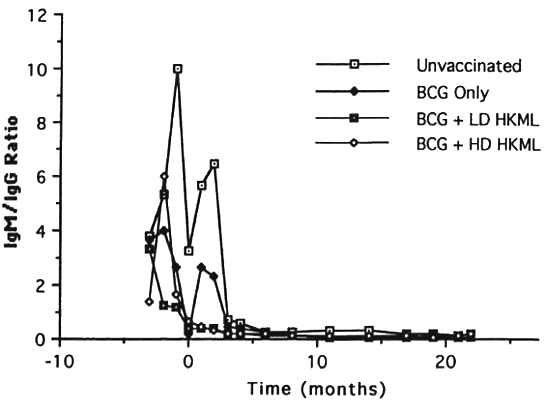
Fig. 9. Longitudinal ratios of IgM:IgG anti-PGL-I antibody isotypes (mean ratio per group).
DISCUSSION
Longitudinal blastogenic responses to lepromin revealed that the initial sensitization to ML antigens peaked within 49 days postvaccination with BCG + HKML. The largest peak response was in the BCG + HD HKML group (SI = 28) followed by the BCG + LD HKML group (SI = 12). Boosting approximately 49 days postvaccination failed to elicit a blastogenic anamnestic respouse; rather, responses rapidly diminished to near baseline by approximately 10 weeks postvaccination. The characteristics of the postboostcr blastogcnic response patterns suggested the possibility of antigeninduced blastogcnic unresponsiveness after the initial responses to vaccination.
Challenge with live ML approximately 10 weeks after vaccination (approximately 3 weeks postboosting) resulted in further diminution of blastogcnic responses to ML antigens to baseline levels in the BCG + HKML groups. Thereafter, aside from intermittent, small, statistically significant increases in Sis in the various vaccinated groups, until approximately 24 months PI, there was no indication of potent ML antigen blastogcnic anamnestic recognition/response by blood MNC. At 24 months PI, small significant peaks of blastogcnic rcsponsivity transiently appeared in the BCG + LD HKML and the BCG + HD HKML groups. Nonetheless, in spite of this apparent indication of a lack of a strong immunologic recognition/response by this criterion, RM croups vaccinated with BCG alone, BCG + LD HKML and BCG + HD HKML all showed significant clinical protection, as we have previously reported (12). Therefore, the blastogcnic response to ML antigens after priming of blood MNC by vaccination/boosting with BCG or BCG + HKML is a misleading indicator of the immunologic activity that controls susceptibility/resistance to clinical leprosy.
During the period of longitudinal blastogenic observation, clinical leprosy was progressing significantly in essentially all of the unvaccinated, ML-challenged group, but only in 5 of the BCG-only vaccinated and in 3 of 20 of the BCG + LD and HD HKML groups (,2). Although there were significant differences in the clinical results, the ML-specific blastogcnic response profiles were surprisingly similar among the four groups after ML-challenge. Protection by BCG or BCG + HKML vaccination appears to be notable more due to a lack of a vigorous, sustained, systemic MNC response than to the expected strong, anamnestic, blastogenic response to ML antigens in the clinically protected groups after challenge with live ML. The exact effects on the results of this study of the combined i.v. and i.e. routes and relatively high doses of ML used for inoculation compared to other possible doses and routes are not known with certainty, but the combined i.v./i.c. routes have previously proven to be the most effective in producing advancing disease in unvaccinated SMM (15,17,19).
We have previously reported that no detectable PGL-I antigen was present in the serum of individual RMs in any of the four ML-challenged (unvaccinated and three vaccinated) groups, although clinical leprosy progressed in the unvaccinated group and a percentage of the vaccinated animals l2). This suggests that the systemic bacterial load was low in these animals, whether protected from clinical leprosy or not. Similar PGL-I antigen data from SMM involved in these same vaccine studies and inoculated by the same routes with similar numbers of ML from the same source showed highly significant levels of PGL-I, differing in amounts between groups, in essentially all ML-challenged animals for the duration of the study (l2). SMM are naturally more susceptible to (multibacillary) leprosy than (paucibacillary prone) RM (12,17-18). Thus, unvaccinated RM are able to maintain relative clearance of ML antigens from the circulation in the face of progressive clinical leprosy, suggesting that some degree of immune containment of ML growth is operative at the paucibacillary end of the leprosy spectrum even though there was a lack of sustained, specific, blastogenic responsiveness of blood MNC to ML antigens.
Longitudinal blastogcnic profiles similar to those described for lepromin, but lower in magnitude, were observed in response to Rces soluble ML antigen and, lowest of all in magnitude, to ML rlO-kDa protein over the same time course. These antigens therefore appear to be good indicators of antilcprosy cellular immunity but, not surprisingly, the more refined the antigen preparation, the lower the magnitude of the response.
Longitudinal observations of the blood lymphocyte subsets provided interesting insights pertaining to the dynamics of the systemic cellular-immune compartment over the course of this study. Changes in the numbers of T-cell subsets with time in the unvaccinatcd, unchallenged (normal) control groups represent a natural phenomenon, perhaps related to seasonal biorhythms ::). These types of fluctuations mandate that normal control groups be included in any longitudinal immunologic study of primates.
Significant increases were observed in the numbers of all subsets studied (CD4+, CD4+CD29+, CD4+CD45RA+ and CD8+) in all three vaccinated, ML-challcnged groups compared to the unvaccinatcd, MLchallcnged group, with the exceptions that: 1) there was no significant increase in numbers of any subset in BCG-only vaccinated animals, and yet this group was protected from clinical disease by 70%; and 2) the changes seen in the CD4+CD29+ and the CD4+CD45RA+ subsets were not significant in the BCG + HD HKML (or BCGonly) group.
The observed increases in cell numbers were progressive with time after infection with some fluctuations, at least over the first 12 months. There was a progressive increase over time in the CD4+ (helper) subset that platcaucd after approximately 12 months in both BCG + HKML-vaccinated, ML-challcnged groups. We have previously noted the importance of blood CD4+ lymphocytes in RM immune responses to ML (13). The CD4+ subset is known to contain cells that are normally involved in positive blastogcnic responses to specific antigens (26). The observation of increasing numbers of this subset in the face of diminishing specific blastogcnic responsivity to ML antigens in the BCG + HKML-vaccinated groups adds suggestive evidence of some form of negative feedback for systemic ML-spccific blastogcnic responses. This result is consistent with the observation that subsets with the suppressor (CD8+) phenotypc increased progressively in the BCG + HKML-vaccinated groups simultaneously with increasing numbers of the helper phenotype.
Beginning at month 8 and continuing through month 21 PI, significant increases were observed in the CD4:CD8 (helper : suppressor) ratio in the BCG + HKML-vaccinated (but not the BCG-onlγ-vaccinated) groups. This relative increase in CD4+ T cells is consistent with protection in the BCG + HKML-vaccinated groups compared to the uninoculated group, but no similar correlation was seen in the protected BCG-only vaccinated group.
The picture that emerges from a consideration of these T-cell subset dynamics is that vaccinated RM, especially those receiving BCG + HKML, were able to maintain higher numbers of the cell types known to contain subsets that are involved in both immunostimulation and immunosuppression in conjunction with protection from clinical disease in the face of decreasing, systemic blastogenic responses to ML antigens in vitro. The results suggest that both enhancer and suppressor arms of the cellmediated immune (CM1) apparatus are involved, presumably in a balanced manner, in successful elimination of living ML from an infected host.
The results of longitudinal ELISA monitoring of IgG versus IgM anti-PGL-I serum antibody responses were in accord with our previous suggestions that IgG responses correlate with protection and IgM responses with susceptibility to leprosy (15,16,18,19). The IgM : IgG ratio after ML-challenge was greatest in the unvaccinatcd (most susceptible) group > the BCG-only vaccinated group (70% protected) > the BCG + HKML (84% protected) group (l2). We do not yet know the basis for this association between anti-PGL-I antibody isotype and leprosy susceptibility, but it is consistent and has been verified in multiple studies in our laboratory (15,16,18,19). Others have noted similar anti-PGL-I IgG and IgM ratio relationships in patients undergoing erythema nodosum leprosum (ENL) or reversal reactions (27). We have described an SMM with experimental borderline lepromatous leprosy and intraneural ENL which maintained an IgG anti-PGL-I antibody pattern for the duration of the ENL episode, but reverted to higher IgM : IgG ratios during the later stages of disease (2). These types of reactional episodes are regarded as manifestations of episodic enhancement of anti-ML CMI.
It is probable that a critical balance between the CMI and the humoral compartments of the immune system, maintained within certain tolerances by relative amounts and types of cytokines produced by various MNC subsets, are required for the successful elimination of living ML (13). In general, more resistant animals maintain an IgM: IgG ELISA ratio <1; individuals which are susceptible to lepromatous forms of leprosy evolve an IgM : IgG ratio >>1 as the disease progresses (19). This IgM/IgG relationship offers an example of a possible subtle effect that may be important in the successful elimination of ML from infected hosts. The critical combination of putative lymphokines may stimulate the production of IgG anti-PGL-I which may play a crucial activation role at the level of the macrophage, for example, in successful immune elimination of ML. Interferon gamma (IFN-γ) is recognized as pivotal in the control of immune responses to ML, and is recognized as important in antibody class switching (20,21,25). TGF-β is also known to be important in immunoglobulin class switching (2'). Thus, the anti-PGL-I IgG:IgM ratio effect could be partially under the control of lymphokines such as IFN-γ and/or TGF-β which are known to be produced by activated CD4+ and CD8+ subsets observed to increase in protected RM in this study (4,21,29).
It remains possible that the antibody response may play no key or direct role in successful protective responses to ML antigens. The antibody profile may be inadvertently controlled by relative amounts of cytokines produced by various subsets that arc directly involved in anti-ML CMI. Additional studies will be necessary to determine the mechanism responsible for the correlation between IgG anti-PGL-I antibody production and resistance to clinical leprosy.
The results show that protection from leprosy correlates positively in some respects with initial postvaccination in vitro (systemic) blastogenic responses of blood MNC, but not after ML-challengc. Vaccination with BCG failed to elicit blastogenic responses or significant changes in T-ccll subset numbers post-ML-challcnge, but was effective in protection. Vaccination with BCG + HD HKML versus BCG + LD HKML produced differing patterns of responses in T-ccll subsets, but were similarly effective in protection. The most consistent correlate to antileprosy protective immunity in this study, as well as our prior studies (2.15,16,18,19), is the parallel between protection and the presence of high levels of serum IgG antibody to PGL-I in comparison to IgM anti-PGL-I. Thus, multiple overlapping pathways appear to be involved in the complex interrelationships that can contribute to protection from and/or resistance/susceptibility to leprosy.
Acknowledgment. These studies were supported by a grant from the National Institute for Allergy and Infectious Diseases (#AI-19302) and by a grant from the National Center for Research Resources (#RR00164). We are indebted to the following persons for skillful technical assistance and data handling: Cynthia Trygg, Carol Coyne, Valerie Smith, Calvin Lanclos, Eva Pecunia and Doris O'Leary.
REFERENCES
1. ABEL, L., CUA, V. V., OBERTI, J., LAP, v. D., DUE, L. K., GROSSIT, J. and L AGRANGE, P. H. Leprosy and BCG in Southern Vietnam, (letter) Lancet 335 (1990) 1536.
2. BASKIN, G. B., GORMUS, B. J., XU, K., MARTIN, L. N., WOLF, R. H., C ANTRELL, C, PEZESHKPOUR, G. H., WALSH, G. P., MAI.ATV, R. and MEYERS. W. M. Experimental borderline lepromatous leprosy with intraneural erythema nodosum leprosum in a mangahey monkey (Cercocelm.s. atys). Int. J. Lepr.59 (1991) 618-623.
3. BECHELLI, L. M., LwiN, K., GALLEGo-GARBOSA, P.G., GY1, M. M., UEMURA, K., SUNDARESAN, T., TAMODONG, C., SANSARRITQ, H. and WALTER, J. BCG vaccination of children against leprosy: nineyear tindings of the controlled WHO trial iaBurma. Bull. WHO 51 (1974) 93-99.
4. BOEHM, U., KLAMP T., GROOT, M. and HOWARD, J. C. Annual' Reviews of Immunology. Palo Alto,California, U.S.A.: Annual Reviews, Inc., 1997, pp. 749-795.
5. CONVIT, J., SAMPSON, C., ZUNIGA, M., SMITH, P.G., PLATA, J., SILVA, J., MOLINA, J., PINARDI, M. E.,13 BLOOM, 13. R. and SALGADO, A. Immunoprophy-lactic trial with comhined Mycobacterium lep-rae/BCG vaccine against leprosy: preliminary re-sults. Lancet 339 (1992) 446-450.
6. DRAPER, P. Protocol 1/79: puritication of M. leprae. Annex 1. Report of the Enlarged Steering Committee meeting, Geneva, 7-8 February 1979.Geneva: World Ilealth Organization, 1979.
7. FINE, P. E. M. BCG vaccination against tubercu-losis and leprosy. Br. Med. Bull. 44 (1988)691-703.
8. FINE, P. E. M. The BCG story: lessons from thepast and implications for the future. Rev. Infecto Dis. 11 Suppl. 2(1989) 5353-S359.
9. FINE, P. E. M. Variation in protection by BCG: implications of and for heterologous immunity [seecommentsl. Lancet 346 (1995) 1339-1345. Published erratum appears in Lancet 347 (1996) 340.
10. FINE, P. E. M., CLAYToN, D., PONNIGHAUS, J. M.and WARNDoRr, D. K. Randomized controlledtrial of single BCG, or combined BCG and killedMycobacterium lepme vaccine for the preventionof leprosy and tuberculosis in Malawi. Lancet 348(1996) 17-24.
11. FINE, P. E. M., PONNIGHAUS, J. M., MAINE, N.,CLARKSON, J. A. and BLISS, L. Protective efticacyof BCG against leprosy in northern Malawi.Lancet 2 (1986) 449-502.
12. GORMUS, B. J., BASKIN, G. B., Xu, K., Botim, R.P., MACK, P. A., RATTERREE, M. S., COO, S.-N., MEYERS, W. M. and WALSH, G. P. Protective im-munization of monkeys with BCG or BCG plusheat-killed Mycobacterium leprae: clinicai results.Lepr. Rev. 69 (1998) 6-23.
13. GORMUS, B. J., MuRRHEY-CoRB, M., MARTIN, L.N., BASKIN, G. B., MACK, P. A., Xu, K., RATTEREE, M. S., GERONE, P. J., ScoLLARD, D. M. and Gins,T. P. Impaired responses to Mycobacterium lep-me antigens in rhesus monkeys experimentally in-oculated with simian immunodeficiency virus and M. leprae. Lepr. Rev. 69 (1998) 24-39.
14. GORMUS, B. J., MuRPIIEY-CORB, M. A., MARTIN, L. N., ZOANG, J. Y., BASKIN, G. B., TRYGG, C. B.,WALSH, G. P. and MEYERS, W. M. Interactions be-tween simian immunodeficiency virus and Mycobacteriulm leprae in experimentally inoculzitedrhesus monkeys. J. Infect. Dis. 160 (1989)405-413.
15. GORMUS, B. J., OHASHI, D. K., OHKAWA, S.,WALSH, G. P., MEYERS, W. M., BRENNAN, P. J. and TRYGG, C. B. Serologic responses to Mycobacteriulm leprae-specilie phenolic glycolipid I anti-gen in sooty nizingahey monkeys with experimen-tal leprosy. Int. J. Lepr. 56 ( 1988) 537-545.
16. GORMUS, B. J., X11, K. Y., ALFORD, P. L., LEE, D. R.,HUBBARD, G. 13., EICHBERG, J. W. and MEYERS, W.M. A serologic study of naturally acquired leprosyia chimpanzees. Int. J. Lepr. 59 (1991) 450-457.
17. GORMUS, B. J., Xu, K., BASKIN, G. 13., MARTIN, L.N., BOFINI, R. P., BLANCHARD, J. L., MACK, P. A.,RATTERREE, M. S., MCCLURE:, H. M., MEYERS, W.M. and WALSH, G. P. Experimental leprosy inmonkeys. I. Sooty mangahey monkeys: transmis-sion, susceptibility, clinica! and pathologiczil tind-ings. Lepr. Rev. 66 (1995) 96-104.
18. GORMUS, 13. J., Xu, K., BASKIN, G.13., MARTIN, L.N., Boftm, R. P., JR., BLANCHARD, J. L., MACK, P.A.,M. S., MEYERS, W. M. and WALSH, G. P. Experimental leprosy in rhesus monkeys:transinission, susceptibility, clinicai and immuno-logical findings. Lepr. Rev. 69 (1998) 235-245.
19. GORMUS, 13. J., Xu, K., COO, S.-N., BASKIN, G. B.,Botim, R. P., MARTN, L. N., BLANCHARD, J. L.,MACK, P. A., RATTERREE, M. S., MEYERS, W. M.and WALSH, G. P. Experimental leprosy in mon-keys. II. Longitudinal serological observations insooty mangabey monkeys. Lepr. Rev. 66 (1995)105-125.
20. KAPLAN, G. and COHN, Z. A. Leprosy and cell-mediated immunity. Curr. Opin. Itunionol. 3(1991) 91-96.
21. KEMENY, D. M., NOBLE, A., HOLMES, B. J. and DIAS-SANCHEZ, D. Immune regulation: a new rolefor the CD8+ T cell. Immunol. Today 15 (1994)107-110.
22. MARTIN, L. N., GORMUS, B. J., WOLF, R. H.,GERONE, P. J., MEYERS, W. M., WALSH, G. P., BINFORD, C. H., HADFIELD T. L. and SCHLAGEL, C. J. Depression of lymphocyte responses to rnitogensin mangaheys with disseminated experimentalleprosy. Cell. Immunol. 90 (1985) 115-130.
23. MARTIN, L. N., MURPHEY-CORB, M., SOIKE K. F., DAVISON-FAIRBURN, B. and BASKIN, G. B. Effects of initiation of 3'-azido, 3'-deoxythymidine (Zidovudine) treatment at different times atter infec-tion of rhesus monkeys with simian immunodeticiency virus. J. Infect. Dis. 168 (1993) 825-835.
24. MARTIN, L. N., SOIKE, K. F., MURPHEY-CORB M., BOHM, R. P., ROBERTS, E. D., KAKUK, T. J., THAISRIVONGS, S., VIDMAR, T. J., RUWART, M. J., DAVIO, S. R. and TARPLEY, W. G. Effects of U-75875, a peptidomimetic inhibitor of retroviral proteases,on simian immunodeticiency vinis infection inrhesus monkeys. Antimicrob. Agents Chemother.38 (1994) 1277-1283.
25. MISRA, N., MURTAZA, A., WALKER, B., NARAYAN, N. P. S., MISRA, R. S., RAMESH V., SINGH, S.,COLSTON, M. J. and NATH, I. Cytokine prolile ofcirculating T cells of leprosy patients rellectsboth indiscriminate and polarized T-helper subsets: T-helper phenotype is stable and uninllu-enced by related antigens of Mycobacterium leoprae. Immunology 86 (1995) 97-103.
26. MULLER, D. L., JENKINS, M. K. and SCHWARTI, R.H. Animal Reviews of Immunology. Palo Alto,California, U.S.A.: Anona! Reviews, Inc., 1989,pp. 445-480.
27. SAHA, K., CHATTOPADHYA, D., KASHYUP, A.,AGARWAL, U. and CHAKRAISARTY, A. K. Enhanced response of serum IgG class of anti-PGL-I anti-bodies in leprosy patients during onset and fol-lowing clinicai remission of type 1 and type 2 re-actions. Int. J. Lepr. 63 (1995) 105-109.
28. STANLEY, S. J., HOWARD, C., STONE, M. M. and SUTHERLAND, I. BCG vaccination of childrenagainst leprosy in Uganda: final results. J. Hyg. 87(1981) 233-248.
29. STAVNEZER, J. Regulation of antibody productionand class switching by TGF-beta. J. Immunol. 154 (1995) 1647-1651.
30. TRIPATHY, S. P. The case for BCG. Ann. Natl. Acad. Med. India 19 (1985) 12-21.
1. Ph.D.; K. Xu, M.D.; Department of Microbiology; Tulane Regional Primate Research Center, 18703 Three Rivers Road, Covington, LA 70433, U.S.A. W. M. Meyers, M.D., Armed Forces Institute of Pathology, Washington, D.C., U.S.A.
2. Ph.D., Department of Microbiology; Tulane Regional Primate Research Center, 18703 Three Rivers Road, Covington, LA 70433, U.S.A. W. M. Meyers, M.D., Armed Forces Institute of Pathology, Washington, D.C., U.S.A.
3. D.V.M., Department of Pathology; Tulane Regional Primate Research Center, 18703 Three Rivers Road, Covington, LA 70433, U.S.A. W. M. Meyers, M.D., Armed Forces Institute of Pathology, Washington, D.C., U.S.A.
4. D.V.M.; Department of Veterinary Sciences, Tulane Regional Primate Research Center, 18703 Three Rivers Road, Covington, LA 70433, U.S.A. W. M. Meyers, M.D., Armed Forces Institute of Pathology, Washington, D.C., U.S.A.
5. D.V.M.; Department of Veterinary Sciences, Tulane Regional Primate Research Center, 18703 Three Rivers Road, Covington, LA 70433, U.S.A. W. M. Meyers, M.D., Armed Forces Institute of Pathology, Washington, D.C., U.S.A.
6. D.V.M., Department of Veterinary Sciences, Tulane Regional Primate Research Center, 18703 Three Rivers Road, Covington, LA 70433, U.S.A. W. M. Meyers, M.D., Armed Forces Institute of Pathology, Washington, D.C., U.S.A.
7. Walsh, Ph.D., American Leprosy Foundation, Rockville, MD, U.S.A.
Reprint requests to Dr. Gormus at the above address or FAX 1-504-893-1352; e-mail: gormus@tpc.tulane.edu
Received lor publication on 25 February 1999.
Accepted for publication in revised form on 4 November 1999.