- Volume 65 , Number 4
- Page: 469–76
Evaluation of methods for isolation of dna f rom slowly and rapidly growing mycobacteria
ABSTRACT
Mycobacteria generally have thick cell walls and contain large amounts of lipid, making them resistant to DNA extraction. Five methods, namely, extensive enzymic digestion method (Ml), 2-min mechanical glass-bead disruption method (M2), thermal shock method (M3), modified conventional enzymic digestion method (M4), and manual disruption with modified conventional enzymic digestion method (M5), were used to compare their effectiveness and simplicity in extracting DNA f rom slowly growing mycobacteria (Mycobacterium leprae, M. lepraemurium and M. bovis BCG), and a rapidly growing mycobacterium (M. phlei). The highest DNA yield was obtained by M2 f rom M. lepraemurium which produced 2.82 pg DNA/mg wet weight of cells, representing a theoretical yield of 78%. M3 gave the lowest DNA yield; 0.01 pg DNA/mg wet weight of cells of M. lepraemurium was obtained. M4, in which proteinase K was used, is more effective than Ml, in which subtilisin and pronase were used. M5 yielded a higher amount of DNA, but it required more manipulations to extract DNA as compared to M4. Extraction of DNA of M. leprae f rom nude mice is more difficult than that of M. leprae f rom armadillos by all of the methods used. These results suggest that the biosynthetic capabilities of these two forms of M. leprae may vary, depending on their cultural conditions and/or strain differences. Our results have shown that both M2 and M4 are the simplest, most effective and timesaving methods which are suitable for every routine laboratory to extract DNA f rom slowly and rapidly growing mycobacteria.RÉSUMÉ
Les mycobactéries ont généralement des parois cellulaires épaisses et contiennent de grandes quantités de lipides, les rendant résistantes à l'extraction de I'ADN. Cinq méthodes-la digestion enzymatique extensive (Ml), la dislocation mécanique durant deux minutes sur des perles de verre (M2), le choc thermique (M3), la digestion enzymatique conventionnelle moditiée (M4) et la dislocation manuelle avec une digestion enzymatique conventionnelle modifiée (M5)-ont été utilisées pour comparer leur efficacité et leur simplicité pour extraire l'ADN de mycobactéries à croissance lente (Mycobacterium leprae, M. lepraemurium et le BCG de M. bovis), et une mycobactéric à croissance rapide (M. phlei). La teneur la plus élevée en ADN a été obtenue par la M2 à partir de M. lepraemurium qui a produit 2,82 ug d'ADN/mg de poids humide des cellules, ce qui représente une teneur théorique de 78%. M3 a donné la teneur la plus faible en ADN; on a obtenu 0,01 ug d'ADN/mg de poids humide des cellules de M. lepraemurium. M4, dans laquelle la protéinase K fut utilisée, est plus efficace que Ml, dans laquelle la subtilisine et la pronase furent utilisées. M5 a donné une quantité plus élevée d'ADN, mais a nécessité plus de manipulations pour extraire l'ADN, par comparaison avec M4. L'extraction de l'ADN de M. leprae provenant de souris nues est plus diffeile que son extraction à partir de M. leprae provenant de tatous, et ceci pour toutes les méthodes utilisées. Ces résultats suggèrent que les capacités biosynthétiques de ces deux formes de M. leprae peuvent varier en fonction de leurs conditions de culture et/ou de différences d'espèce. Nos résultats ont montré que M2 et M4 sont toutes les deux les méthodes les plus simples, les plus efficaces et les plus rapides qui soient adaptées à tout laboratoire de routine pour extraire l'ADN de mycobactéries à croissance lente et rapide.RESUMEN
Las micobactcrias en general, tienen gruesas paredes celulares y contienen grandes cantidades de lípidos que dificultan la extracción de su DNA. En este estudio se compararon 5 métodos de extracción del DNA de micobacterias de lento crecimiento (Mycobacterium leprae, M. lepraemurium y M. bovis, BCG) y de una micobacteria de crecimiento rápido (M. phlei). Los métodos fueron un método de digestión enzimática extensa (MI), un método de rompimiento mecánico por 2 min con perlas de vidrio (M2), un método de choque térmico (M3), un método modificado de digestión enzimática convencional (M4), y un método de rompimiento manual combinado con digestión enzimática (M5). El mayor rendimiento de DN A se obtuvo con el M2 aplicado a M. lepraemurium (2.82 ug de DNA por mg de peso húmedo). Con esta bacteria, el M3 dio el menor rendimiento (0.01 ug por mg de peso húmedo). El M4, en el cual se usa proteinasa K, es más efectivo que el MI en el que se usan subtilisina y pronasa. E1M5 produjo una mayor cantidad de DNA pero se requiere de más manipulación que en el M4. La extracción de DNA del M. leprae propagado en el ratón desnudo es más difícil que la extracción de DNA del M. leprae cultivado en el armadillo, por todos los métodos usados. Estos resultados sugieren que las capacidades biosintéticas de estas dos formas de M. leprae pueden variar dependiendo de sus condiciones de cultivo y de las diferencias entre las posibles cepas. Nuestros resultados han demostrado que tanto M2 como M4 son los métodos más simples, efectivos y cortos para la extracción de DNA de micobacterias tanto de crecimiento rápido como de crecimiento lento.At present, 71 recognized or proposed species meet the standards for inclusion in the genus Mycobacterium. These species are usually grouped into two major divisions, "rapidly growing" and "slowly growing," based on the time required for visible colonies to appear on a solid medium. The appearance of colonies requires less than 7 days for the rapidly growing species and more than 7 days for the slowly growing species. These divisions are very important clinically and in identification schemes. The slowly growing species may cause disease in humans and animals; whereas the rapidly growing species do not (18,19,20). More than 25 species in the Mycobacterium genus, including M. leprae and M. tuberculosis, may cause disease in humans, and more than six species may cause disease in animals.
Mycobacterial cell walls have been shown to contain type-specific antigenic glycolipids, such as phenolic glycolipids (PGL), glycopeptidolipids (GPL), and trehalose-containing lipo-oligosaccharides (LOS). The compositions of these waxy lipids, mycolic acid-containing cell walls vary depending upon the species of Mycobacterium (1,2,7,21). Various methods have been described for the extraction of DNA from mycobacteria (3.6,9,22) The critical step is lysis of the cells to release DNA, while avoiding its degradation or shearing. Due to the cell-wall thickness, mycobacteria are resistant to enzyme lysis by the commonly described methods. The ability to identify and to type mycobacteria from environmental and clinical sources is limited by the lack of a simple, rapid and efficient method of DNA preparation by which many samples can safely be processed simultaneously (22). Since the morphology and the biosynthetic capabilities of mycobacteria vary, depending on cultural conditions and strain differences within a species, the most reliable method of identification would be at the DNA level (15.19).
Although research in molecular biology and recombinant DNA technology has advanced markedly in recent years, the lack of a simple and efficient method for extracting DNA from mycobacteria has lagged behind the need of research. During this study, five methods were used to extract DNA from slowly growing mycobacteria such as M. leprae and M. lepraemurium, the causative agents of human and murine leprosy, respectively, as well as M. bovis BCG, an etiologic agent of tuberculosis. M. plilei, a nonpathogen and rapidly growing mycobacterium, was used as a control. The quantity and the quality of these DNAs were analyzed by electrophoresis and restriction endonuclease digestion.
MATERIALS AND METHODS
Sources and growth of mycobacteria
M. bovis BCG substrain Laval was kindly provided by Dr. P. Rousseau (BioVac, Institut Armand-Frappier, Laval, QC, Canada), after being grown on Sauton medium. M. phlei was grown on the Lowenstein-Jensen medium at 37ºC for a period of 5-7 days (15). M. leprae were obtained from infected foot pads of nude mice maintained in our laboratory (12). M. leprae from infected armadillo liver tissue was a gift kindly provided by Dr. A. M. Dhople, Florida Institute of Technology, Melbourne, Florida, U.S.A. M. lepraemurium Hawaiian strain used in this study was obtained from Dr. P. Greenburg (La Roche Research Laboratory, Nutley, New Jersey, U.S.A.). The strain was maintained in C3H mice by subcutaneous serial passages every 4-6 months in our laboratory (13).
Reagents
Reagents used for isolation of M. leprae and M. lepraemurium cells from infected animals were those recommended by Draper (10) and Clark-Curtiss, et al . (8). Buffer A contained 150 mM NaCl, 1 mM MgS04 and 15mM HEPES (N-2-hydroxyethylpiperazine-N'-2-ethanesulfonic acid), pH 7.2. Buffer B contained 150 mM NaCl, 200 mM Tris buffer, and 1 mM MgS04, pH 7.2. Buffer C contained 1 ml 10%Tween80, 0.2 ml of 10% 2-(N-morpholino)ethanesulfonic acid (MES), and 100 ml distilled water, pH was adjusted to 7.2. All three buffers contained 1 mM benzamidine as an inhibitor of animal cellular proteases. The 30% Percoll gradient contained 30 ml Percoll (Sigma Chemical Co., St. Louis, Missouri U.S.A.) and 70 ml buffer C.
Reagents for DNA extraction and preservation were described by Sambrook, et al. (17). NET buffer contained 100 mM NaCl, 10 mM EDTA and 10 mM Tris-HCl, pH 7.5. TE buffer contained 1 mM EDTA and 10 mMTris-HCI, pH 7.5.
The enzymes, such as proteinase K, pronase, lysozyme, DNase, and RNase A were purchased from Boehringer Mannheim (Laval, Canada); the other enzymes, such as subtilisin (protease VIII) and colla-genase were purchased from Sigma; Pfu DNA polymerase was purchased from Stratagene (La Jolla, California, U.S.A.); Pst I was purchased from Pharmacia Biotech, (Baie d'Urfe, Canada).
Isolation and preparation of mycobacterial bacillary suspensions
Isolation of M. leprae bacilli from infected nude mouse foot pads and preparation of bacillary suspensions. The method for quantitative recovery of M. leprae cells from infected nude mouse foot pads was adapted from a method developed by Franzblau and Hastings (11) with some modifications. Briefly 10 g of foot pads were surface decontaminated by using ultraviolet (UV) irradiation, 1% iodine and 70% ethanol. Foot pads were then cut into small pieces with a pair of scissors and triturated thoroughly in a mortar in buffer A. The mixture was passed through a sterile nylon filter into a sterile 250-ml beaker to remove tissue strands, and then the filtrate was centrifuged (Beckman J21 B) at 1200 x g x 10 min at 4ºC to remove the tissue debris. The tissue debris was triturated and filtered through a nylon filter once more as described above to further recover the bacilli. The pooled filtrate was centrifuged at 10,000 x g x 15 min at 4ºC. The resulting supernatant was carefully removed, without disturbing the pellets, and discarded. The pellets were suspended in 30 ml NET buffer.
Enzymatic digestion of foot-pad tissues and collagen was accomplished by adding 4 mg of collagenase to the flask containing 30 ml of the homogenized mixture in the NET buffer. The flask was incubated at 37ºC with gentle shaking for 1 hr. A solution containing 0.5% SDS and 0.1 mg/ml of proteinase K was added and incubated at 56ºC for 1 hr. Thereafter, 50 µg/ml of DNase was added to the mixture and further incubated for 1 hr to digest nude mouse DNA. The mixture was then transferred into sterile tubes and centrifuged at 10,000 x g x 15 min. The supernatant fractions were carefully removed. The pellets were washed once with buffer B by centrifugation at 10,000 x g x 15 min. The resulting pellets were suspended in 12 ml of buffer C using a Pyrex tissue grinder.
Further purification of the bacilli in 12 ml of buffer C was accomplished by 30% Percoll gradient density separation (16). The suspended cells in 12 ml of buffer C were equally transferred to two sterile Beckman screw-cap centrifuge tubes. To each tube, 24 ml of 30% Percoll were added. The tubes were centrifuged at 20,000 x g x 1 hr at 5ºC. The bottom layers were taken and washed twice with buffer C, twice with 0.9% NaCl solution, and twice with PBS buffer. Finally, the pellets containing pure M. leprae were frozen at -80ºC until used for DNA extraction.
Isolation of M. leprae bacilli from infected armadillo liver tissues and preparation of bacillary suspensions. The isolation of M. leprae bacilli from infected armadillo liver tissues and preparation of bacillary suspensions were carried out as described by Clark-Curtiss, et al. (8).
Isolation of M. lepraemurium bacilli from infected C3H mice and preparation of bacillary suspensions. The isolation and preparation of M. lepraemurium bacilli from C3H mice lepromas was carried out as described in isolation of M. leprae bacilli from infected nude mouse foot pads.
Preparation of M. bovis BCG bacillary suspensions. M. bovis BCG was grown on the synthetic Sauton medium for 14 days. The bacilli were removed carefully from the surface of the medium. They were washed twice with PBS buffer by centrifugation at 10,000 x g x 10 min, and the pellets were frozen at -80ºC until used for DNA extraction.
Preparation of M. phlei bacillary suspensions. Actively growing cells of M. phlei on the surface of Lowenstein-Jensen medium were carefully removed. They were washed twice with PBS buffer by centrifugation at 10,000 x g x 10 min, and the pellets were frozen at -80ºC until used for DNA extraction.
DNA extraction methods
Five different DNA extraction methods were compared for their ability to provide quality template DNA for endonuclease digestion. Before the DNA extraction, the mycobacterial cells were incubated at 70ºC for 1 hr to minimize any biological hazard. Cells (about 50 mg wet weight) of each mycobacterium were harvested from suspension by centrifugation at 12,000 x g x 5 min, and resuspended in Eppendorf tubes containing 1 ml NET buffer to be used in the following methods.
Intensive enzymic digestion method (M1). The intensive enzymic digestion method was carried out according to Visuvanathan, et al. (23) with modifications. To 1 ml of the cell suspension, 5 mg of subtilicin was added and the mixture was incubated at 37ºC for 18 hr. Then 20 mg of lysozyme was added and the mixture was incubated at 50ºC for 5 hr. Lysis was completed by the addition of 3% SDS and 1.5 mg of pronase. The mixture was further incubated for 20 hr, with the addition of a further 1.5 mg of pronase after 12-15 hr.
Two-min mechanical glass-bead disruption method (M2). The 2-min mechanical glass-bead disruption method was carried out as described by Via and Falkinham (22) and Jacobs, et al. (14) with some modifications. Briefly, the cells (50 mg) were added in a 2-ml screw-cap microcentrifuge tube. One g of 0.1 mm glass beads (Glasperlan; B. Braun Diessel Biotech GmbH, Melsungen, Germany) was added, and the tube was agitated in a shaking bead mill (B. Braun Diessel Biotech) for 2 min.
Thermal shock method (M3). In the thermal shock method, the cells (50 mg) were resuspended in 400 µl of H20 and subjected to a series of heat/cold shocks, alternately boiling (5 min, 100ºC) and snap freezing (5 min, -196ºC in liquid nitrogen). The procedure was repeated six times.
Modified conventional enzymic digestion method (M4). A method described by Ausubel, et al. (5) was used with some modifications. Briefly, lysozyme was added to a final concentration of 2 mg/ml of cell suspension, and the tube was incubated at 37ºC for 1 hr. Then 0.3 mg proteinase K and SDS to a final concentration of 3% were added, and the tube was further incubated at 56ºC for 1 hr.
Manual disruption with modified conventional enzymic digestion method (M5). In the manual disruption with modified conventional enzymic digestion method the cells (50 mg) were placed in a mortar which contained dry ice and glass beads (0.1 g, 0.11 mm, Glasperlan; B. Braun Diessel Biotech). The cells were triturated for approximately 30 min, then transferred to a sterile 1.5 ml Eppendorf tube. The mortar was warmed to room temperature, and the appropriate NET buffer was used to wash any residual cells out of the mortar into the Eppendorf tube. The remaining steps were as described in the M4 method.
DNA purification. RNA elimination and DNA purification in the above five methods were carried out as described below: The lysed sample was centrifuged at 12,000 x g x 10 min in a benchtop microcentrifuge to precipitate the cell debris. The supernatant was recovered, and an equal volume of phenol : isoamyl alcohol : chloroform (25:1:24) was added. The mixture was shaken by hand, and the phases were separated by centrifugation at 12,000 x g x 5 min. The aqueous phase was carefully recovered and an equal volume of chloroform was added. The mixture was shaken and treated as described above. Added to the pooled aqueous phase was 0.1 vol of 3 M sodium acetate, pH 5.2, and 2 vol of 100% ethanol. After thorough mixing, the DNA could often be spooled onto a glass rod, or could be precipitated by centrifugation at 12,000 x g x 20 min. The collected DNA was washed in 70% ethanol, dried briefly, and resuspended in 400 µl NET buffer containing 10 µl/ml RNase A. After a 30-min incubation at 37ºC, 10 µl/ml proteinase K and 0.1 % SDS were added and incubated at 56ºC for another 30 min. The DNA was recovered by centrifugation and retreated with phenol, chloroform, sodium acetate, and ethanol as before. Finally, DNA was dissolved in 100 µl TE buffer and stored at 4ºC for at least 24 hr before use to ensure that it had fully dissolved. DNA concentration was determined spectrophotometrically (17).
Electrophoresis and imaging
DNA samples. Approximately 500 ng DNA of each sample was electrophoresed in a 0.7% agarose gel with 1 x TBE (250 mM Tris, 0.5 mM EDTA, 25 mM boric acid). DNA fragments of known sizes of 1 kb DNA ladder marker (Gibco BRL) were loaded in the same agarose gel. The gels were stained with ethidium bromide (0.5 jUg /ml) and photographed on a UV transil-luminator.
Restriction endonuclease digestion of DNA. Each DNA sample (500 ng) was used in a 10 µl volume to perform the restriction endonuclease digestion, in accordance with the manufacturer's recommendations. The other procedures were the same as described in DNA samples.
RESULTS
Isolation and restriction endonuclease digestion of DNA
The different mycobacteria used in these experiments and the respective DNA yields recovered by different methods are shown in The Table. The highest DNA yield of 2.82 µg/mg wet weight cells was obtained by the 2-min mechanical glass-bead disrur tion method (M2) from M. lepraemuriw while the thermal shock method (M3) ga. the lowest DNA yield of 0.01 µg/mg wet weight cells from M. lepraemurium. These results show that 282-fold more DNA was obtained by M2 compared to M3 for this species. Ml and M3 were the least effective methods for DNA extraction from all the mycobacteria tested. M2 was found to extract DNA most efficiently from in vitro grown M. phlei. Manual disruption with modified conventional enzymic digestion method (M5) yielded comparatively higher yields of DNA from all mycobacteria used than those obtained by the modified conventional enzymic digestion only (M4). It is interesting to note that it is relatively more difficult to extract DNA from M. leprae recovered from foot pads of nude mice than the bacilli isolated from armadillo liver tissues (The Table).
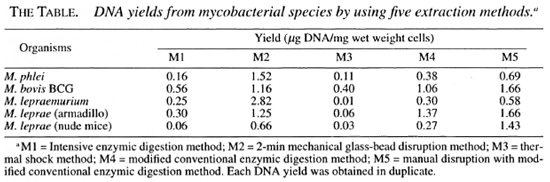
The purified DNAs were separated by electrophoresis on a 0.7% agarose gel to evaluate the quality and the size of DNA fragments (Fig. 1). Every DNA tested exhibited a major band larger that 12 kbp (Fig. 1), and could be completely digested by Pst I (Fig. 2).
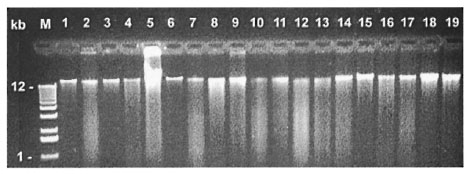
Fig. 1. DNA prepared from five mycobacterial species by using five DNA extraction methods. Five species: M. phlei = lanes I-3; M. bovis BCG = lanes 4-8; M. lepraemurium = lanes 9-12; M. leprae (armadillo) = lanes13-16; M. leprae (nude mice) = lanes 17-19. Five DNA extracting methods: Intensive enzymic digestion method (MI) = lanes 4, 9, 13; 2-min mechanical glass-bead disruption method (M2) = lanes 1, 5, 10, 14, 17; thermalshock method (M3) = lane 6; modified conventional enzymic digestion method (M4) = lanes 2, 7, 11, 15, 18; manual disruption with modified conventional enzymic digestion method (M5) = lanes 3, 8, 12, 16, 19.
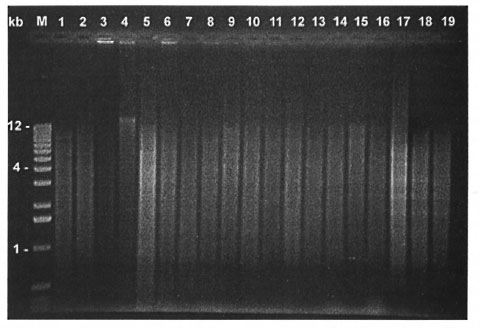
Fig. 2. Same as Figure I, but DNA was digested by Pst I.
DISCUSSION
The extraction of mycobacterial DNA has not been well exploited due to the thick, waxy lipid, and mycolic acid-containing mycobacterial cell walls that are extremely difficult to be lysed by methods routinely used. Visuvanathan, et al. (23) used an extensive enzymic digestion method (Ml) in which subtilisin and pronase were used for isolation of DNA from different bacteria, and reported that this method was also useful for DNA extraction from mycobacteria. Our results have shown that this method (M1) gave much lower yields of DNA from all mycobacteria tested as compared to M2, M4 and M5. In our study, we found that a modified enzymic digestion method (M4), in which the concentrated proteinase K and SDS were used, was quite effective for all rapidly and slowly growing mycobacteria tested (The Table). Contrary to the conclusion of Bollet, et al. (6), who indicated that proteinase K lacks efficacy on most mycobacterial species, we found that proteinase K is more effective than subtilisin and pronase to digest mycobacterial cell walls under the same conditions used in this study, especially for M. leprae derived from both armadillo liver tissues and foot pads of nude mice. It should be emphasized that this is the first study in which M. leprae purified from foot pads of nude mice and armadillo liver tissues have been used to investigate whether or not there could be differences in extracting DNA by various methods. M4 yielded more than three times higher DNA than Ml from both sources of M. leprae. Moreover, M4 takes less than 3 hr, while at least 40 hr are required for DNA extraction by M1.
Both 2-min mechanical glass-bead disruption (M2) and manual disruption with modified conventional enzymic digestion (M5) methods recovered more DNA than any other methods tested. The results in The Table show that M2 recovered the highest quantity of DNA; 2.82 µg of DNA/mg wet weight cells of M. lepraemurium (about 109 bacilli). This represents a theoretical yield of about 78% (4,23). From a practical point of view, when M2 and M5 are compared the former needs special equipment and the latter demands more technical effort to produce reproducible results. Our experience has shown that M4 is the easiest, the least time-consuming, and is an effective method suitable for every laboratory for the extraction of DNA from rapidly and slowly growing mycobacteria. M3 gave the least DNA recovery from M. lepraemurium, yielding only 0.28% of the DNA theoretical yield.
Recently, de Lamballerie, et al. O used "Chelex 100" to extract DNA from M. tuberculosis and M. avium. We have also tried this method which seemed effective for DNA extraction from M. phlei, M. bovis BCG, and M. lepraemurium, but not from M. leprae either recovered from armadillo liver tissues or from the foot pads of nude mice (data not shown). It is possible that the chemical compositions of the cell walls of M. leprae differ from the other mycobacteria tested.
Our results have clearly shown that the yields of DNA of M. leprae isolated from the foot pads of nude mice are slightly lower than those isolated from armadillo liver tissues by all of the methods used in our study (The Table). We have observed from the stained preparations that the bacilli from the liver tissues of armadillos are generally thinner and longer than those from the foot pads of nude mice under the light microscope (data not shown). The biosynthetic capabilities of mycobacteria may vary depending upon cultural conditions and strain differences.
Acknowledgment. We are thankful to Dr. P. Rousseau for providing us with M. bovis BCG, and to Dr. R. Morosoli and Mr. S. Durand for help in the development of Method 2 used. We are also grateful to Mr. F. Bizouarn for his technical assistance and Dr. F. Sharek for helpful discussions and critical reading of the manuscript.
This study was generously supported by Le Secours aux Lépreux, Canada, Inc.; by the Military and Hospitaller Order of Saint Lazarus of Jerusalem, Canada; and by the German Leprosy Relief Association, Wurzburg, Germany.
REFERENCES
1. ALUGUPALLI, S., LANEELLE, M. A., LARSSON, L. and DAFFE, M. Chemical characterization of the ester-linked 3-hydroxy fatty acyl-containing lipids in Mycobacterium tuberculosis. J. Bacteriol. 177 (1994) 4566-4570.
2. ALUGUPALLI, S., PORTALES, F. and LARSSON, L. Systematic study of the 3-hydroxy fatty acid composition of mycobacteria. J. Bacteriol. 176 (1994) 2962-2969.
3. ANDERBERG, R. J., STRACHAN, J. A. and CAN-GELOSI, G. A. Purification of DNA from Mycobacterium species without sonification or phenol. Biotechniques 18 (1995) 217-219.
4. ATHWAL, R. S., DEO, S. S. and IMAEDA, T. Deoxyribonucleic acid relatedness among Mycobacterium lepraemurium, Mycobacterium leprae, and selected bacteria by dot blot and spectrophotometric deoxyribonucleic acid hybridization. Int. J. System. Bacteriol. 34 (1984) 371-375.
5. AUSUHEL, F. M. Current protocols in molecular biology. In: Short Protocols in Molecular Biology; A Compendium of Methods from Current Protocols in Molecular Biology. 2nd edn. Ausubel, F. M., Brent, R., Kinston, R. E., Moore, D. D., Seidman, J. G., Smith, J. A. and Struhl, K., eds. Brooklyn, New York: Greene Publishing Association, Inc. and John Wiley & Sons, Inc., 1992, 99. 2.4.1-2.4.5
6. BOLLET, C, GEVAUDAN, M. J., DE LAMBALLERIE, X., ZANDOTTI, C. and DE MICCO, P. A simple method for the isolation of chromosomal DNA from gram-positive or acid-fast bacteria. Nucl. Acids Res. 19(1991) 1955.
7. BRENNAN, P. J., CHATTERJEE, D., FUJIWARA, T. and CHO, S.-N. Leprosy-specific neoglycoconjugates; synthesis and application to serodiagnosis of leprosy. Methods Enzymol. 242 (1994) 27-37.
8. CLARK-CURTISS, J. B., JACOBS, M. A., DOCHERTY, W. R., RrTCHLE, L. R. and CURTISS, R., III. Molecular analysis of DNA and construction of genomic libraries of Mycobacterium leprae. J. Bacteriol. 161 (1985)1093-1102.
9. DE LAMBALLERIE;, X., ZANDOTTI, C, VIGNOLI, C, BOLLET, C. and DE MICCO, P. A one-step microbial DNA extraction method using "Chelex 100" suitable for gene amplification. Res. Microbiol. 143(1992)785-790.
10. DRAPER, P. Cell walls of Mycobacterium leprae. Int. J. Lepr. 44(1976)95-98.
11. FRANZBLAU, S. G. and HASTINGS, R. C. In vitroand in vivo activities of macrolides against My-cobacterium leprae. Antimicrob. Agents Chemo-ther. 32 (1988) 1758-1762.
12. ISHAQUE, M. and STICHT-GROH, V. Investigationsinto the growth of Mycobacterium leprae in amedium with palmitic acid under different gaseousenvironments. Microbios 75 (1993) 171-179.
13. ISHAQUI:, M., Tocol.A, D. and STtcttT-GRoIt, V. α-Ketoglutarate dehydrogenase in the in vitrogrown Mycobacterium lepraemurium. Int. J. Lepr.62 (1994) 399-403.
14. JACOBS, W. R., JR., RALPANA, G. V., GIRII.LO, J. D., PASCOPELLA, L., SNAPPER, S. B., LIDANit, R. A., JONES, J., BARLETTA, R. G. and BLOOM, B. R. Genetic system for mycobacteria. Methods Enzymol.204 (1991) 537-555.
15. PATEL, R., RVACII, J. T. and MOUNTS, P. Isolationand restriction endonuclease analysis of mycobacte-rial DNA. J. Gen. Microbiol. 132 (1986) 541-551.
16. RODDE, C., MOHAMED, A. A. F., LUESSE, H. G. and KAZDA, J. Improved method for purification of Mycobacterium leprae from armadillo tissues. Int.J. Lepr. 60 (1992) 277-278.
17. SAMBROOK, J., FRITSCH, E. F. and MANIATIS, T.Molecular Cloning: A Laboratory Manual. Cold Spring Harbor, New York: Cold Spring HarborLaboratory Press, 1989, pp. E5-E7.
18. SHINNICK, T. M. and Goon, R. C. Mycobacterialtaxonomy. Eur. J. Clin. Microbiol. Infect. Dis. 13(1994) 884-901.
19. SHINNICK, T. M. and Goon, R. C. Diagnostic my-cobacteriology laboratory practices. Clin. Infect.Dis. 21 (1995) 291-299.
20. SPINGER, B., STocKNIAN, L., TESCHNER, K.,ROBERTERS, G. D., and BOETTGER, E. C. Two-lab-oratory collaborative study on identification ofmycobacteria: molecular versus phenotypic meth-ods. J. Clin. Microbiol. 34 (1996) 296-303.
21. SUTCLIFFE, L. and RUSSELL, R. R. B. Lipoproteinsof gram-positive bacteria. J. Bacteriol. 177 (1995)1123-1129.
22. VIA, L. E. and FALKINHAM, J. 0., III. Comparisonof methods for isolation of Mvcobacterium aviumcomplex DNA for use in PCR and RAPD. J. Mi-crobiol. Methods 21 (1995) 151-161.
23. VISUVANATHAN, S., Moss, M. T., STANFORD, J. L.,HERMON-TAYLOR, J. and MCFADDEN, J. J. Simpleenzymic method for isolation of DNA from di-verse bacteria. J. Microbiol. Methods 10 (1989)59-64.
1. Ph.D.; Centre de Recherche en Microbiologie Appliquée, Institut Armand-Frappier, Universite du Quebec, 531 Boulevard des Prairies, Laval, Quebec H7N 4Z3, Canada.
2. Ph.D., Professor, Centre de Recherche en Microbiologie Appliquée, Institut Armand-Frappier, Universite du Quebec, 531 Boulevard des Prairies, Laval, Quebec H7N 4Z3, Canada.
Received for publication on 6 January 1997.
Accepted for publication in revised form on 17 September 1997.
Reprint requests to Professor Ishaque at the above address or FAX 514-686-5501.