- Volume 64 , Number 4
- Page: 409–16
Detection of M. leprae by gene amplification; combined ethidium-bromide staining and probe hybridization
ABSTRACT
Biopsy and skin-scraping specimens f rom 130 leprosy cases across the disease spectrum (56 TT/BT/I, 73 BB/BL/LL, and 1 neuritic case) and 50 healthy contacts were studied to assess the application of gene amplification. The nucleic acids f rom these clinical specimens were extracted by an integrated freeze-thawing-optimized lysozyme-/proteinase-k treatment-purification and fractionation procedure. The nucleic acids f rom cultured organisms were isolated by the stepwise procedure earlier standardized at this laboratory. Gene amplification for a 360-bp fragment of the 18kDa protein gene was carried out using primer and the procedure described by its developers, and a 360-bp fragment on Southern blot was taken as the yardstick of positivity. The polymerase chain reaction product was analyzed by electrophoresis, ethidium-bromide (EB) staining, and blot (B) hybridization. Overall sensitivity ranged f rom 71% in specimens with undetectable acid-fast organisms to 100% in specimens with demonstrable acid-fast bacilli. A positivity of 73% in TT/BT/I specimens and 93% in BB/BL/LL specimens was observed. Four combinations were discerned: EB+, B+ (71%); EB-suspicious, B+(14%); EB-, B+ (3%) and EB-, B- (12%). By combining the blot hybridization with EB staining, the sensitivity could be significantly improved as compared to EB staining alone. The test was found to be absolutely specific by the absence of any false positivity in control specimens as well as with purified DNAs f rom mycobacterial as well as non-mycobacterial organisms grown f rom these specimens. It is recommended that for optimum sensitivity and specificity both EB staining and blot hy- bridization should be done.RÉSUMÉ
Des biopsies et des échantillons de frottis cutanés de 130 malades de la lèpre à travers tout le spectre de la maladie (56 TT/BT/I, 73 BB/BL/LL, et 1 cas névritique) et 50 contacts en bonne santé ont été étudiés pour évaluer l'application de l'amplification de gènes. Les acides nucléiques de ces échantillons cliniques ont été extraits par un procédé intégré et optimisé de geldégel, traitement au lysozyme-/proteinase-k, purification et fractionnement. Les acides nucléiques des organismes cultivés ont été isolés par la procédure en étapes standardisée pécédemment dans ce laboratoire. L'amplification génique d'un fragment de 360 bp de la protéine de 18-kDa a été réalisée à l'aide d'une sonde et de la procédure décrite par ceux qui ont développé la technique, et un fragment de 360 bp au Southern-blot a été pris comme mesure de positivité. Le produit de la réaction de polynierase en chaîne a été analysé par électrophorèse, coloration à l'ethidium-bromide (EB) et blot hybridisation (B). La sensibilité globale allait de 71% pour les spécimens pour lesquels des bacilles acido-résistants n'étaient pas détectables à 100% pour les specimens pour lesquels la présence de bacilles acido-résislants pouvait être démontrée. On a observé une positivité de 73% pour les specimens TT/BT/I et de 93% pour les specimens BB/BL/LL. On a distingué quatre combinaisons: EB+, B+ (71%); EB suspect, B+ (14.0%); EB-, B+ (3%) et EB-, B(12%). En combinant la blot hybridisation et la coloration à l'EB, la sensibilité pouvait être améliorée significativement par rapport à la coloration à l'EB seule. On a trouvé que le test était absolument spécifique, vu l'absence d'aucun faux positif parmi les specimens de contrôle, aussi bien avec les ADN purifiés des mycobactéries qu'avec des organismes nonmycobactériens ayant poussé à partir de ces specimens. Il est recommandé, pour une sensibilité et une spécificité optimales, de faire la coloration à l'EB et la blot hybridization.RESUMEN
Se estudiaron biopsias y raspados de piel de 130 casos de lepra (56 TT/BT/I, 73 BB/BL/LL, I caso neurítico) y de 50 contactos sanos, para valorar la aplicación de la amplificación de genes. Los ácidos nucleicos de estos especímenes se extrajeron y purificaron por un proceso optimizado que incluye congelación y descongelación, y tratamiento con lisozima y proteinasa K. Las ácidos nucleicos de los organismos cultivables se aislaron por un proceso escalonado estandarizado en este laboratorio. La amplificación de un fragmento de 360 bp del gene de la proteína de 18 kl) se llevó a cabo usando el "primer" y el procedimiento descrito por sus inventores. La aparición de un fragmento de 360 bp en el "southern blot" se tomó como la medida de positividad. El producto de la reacción en cadena de la polimerasa se analizó por electroforesis, por tinción con bromuro de etidio (EB), y por hibridización en el "blot". La sensibilidad global varió del 71% en los especímenes sin bacilos détectables, al 100% en los especímenes con bacilos. Se observó una positividad del 73% en los especímenes TT/BT/I y del 93% en los especímenes BB/BL/LL. Se distinguieron 4 combinaciones: EB+, B+(71%); EB-sospechoso, B+ (14%); EB-, B+ (3%) y EB-, B- (12%). La sensibilidad pudo mejorarse de manera significativa combinando la bibridización (B) con la tinción EB. La prueba resultó absolutamente específica ya que no hubo ninguna falsa-positividad en los especímenes control o en los DNA de micobacterias y otros microorganismos cultivables. Para máxima sensibilidad y especificidad se recomiendo que se haga tanto la tinción con EB como la bibridización en "el blot".Leprosy continues to be a major public health problem in India and several parts of the world. Although the number of leprosy cases are decreasing with widespread coverage of multidrug therapy (MDT), yet because of the inability to grow Mycobacterium leprae in vitro the optimum methodologies to diagnose the disease early, to investigate and prevent its transmission, and to improve present-day treatment are yet to be worked out. In the absence of any acceptable in vitro method of cultivation, the criteria for diagnosis are essentially clinical (35) and histopathological (25). These criteria have their limitations, especially in the case of the early atypical forms which are becoming more important now when the proportion of advanced and well-defined types is decreasing. The qualitative as well as the quantitative demonstration of M. leprae or its chemical components (3,30) or antigens (16) in the lesions has been an important approach to establish the diagnosis ("). Bacteriological assessment by estimation of the bacterial index (BI) by the Dharmendra (6) and the Ridley scales (17,24) has been an important approach for confirming the diagnosis of leprosy and for monitoring the treatment. However, most of these techniques are not that sensitive and specific.
The polymerase chain reaction (PCR) described by Mullis and his colleagues (26) is one of the major technological advances in molecular biology which has changed and replaced several conventional strategies and approaches. During the last decade, several gene sequences of M. leprae have been identified and variable sequences-stretches as targets for probes and PCR have been identified. Various PCR techniques for M. leprae have been reported in the recent years, including the amplification of various gene sequences: part of the 65 kDa protein gene (8,23,24), the 36 kDa protein gene (4,9), the 18 kDa gene (7,31,33); the LSRgene (20), rRNA genes (10,34,36) and repetitive sequences (10,34,36). While these techniques have been reported to be fairly sensitive in the initial application reports, there is limited feedback about their application at the clinical level. Crossreactivity (if any) with mycobacterial (27) and non-mycobacterial organisms (28) present in the clinical specimens is important and needs to be investigated in different patient populations from diverse geographical areas. In this communication, our experience of the application of the gene amplification technique targeting 18 kDa gene sequences in specimens from all types of leprosy cases as well as also with the DNA isolated from the organisms isolated from them is being reported.
MATERIALS AND METHODS
Clinical Specimens
Specimens from 130 untreated leprosy cases (untreated or < 3 months' treatment) across the disease spectrum were studied. These included: 56 paucibacillary (PB) leprosy cases [3 tuberculoid (TT); 43 borderline tuberculoid (BT), including 2 relapses; 10 indeterminate (I) and 73 multibacillary (MB) types [22 borderline borderline (BB), 35 borderline lepromatous (BL) and 16 lepromatous (LL) patients]. These specimens included 110 biopsies and 20 skin scrapings. Fifty control specimens (slit-skin smears from 30 contacts of leprosy patients and 20 skin biopsies from healthy controls) were also studied. All of the biopsies and skin scrapings for this study were obtained from the leprosy patients attending the Outpatient Department (OPD) of the Central JALMA Institute for Leprosy; specimens from healthy controls were obtained from this Institute as well as from S.N. Medical College and Hospital, Agra, India. Controls were comprised of individuals who were either healthy contacts of leprosy patients or were attending the clinic for any disease other than a mycobacterial infection.
Biopsies from leprosy patients, healthy controls and contacts. Biopsies from active lesions of leprosy patients were collected under aseptic conditions using local anesthesia. With a scalpel an elliptical area of skin and subcutaneous tissue weighing about 50-100 mg was removed.
Scrapings from lesions and healthy controls. After using proper antiseptic precautions, scrapings as for skin smears were taken from the leprosy lesions as well as healthy skin (17), collected in peptone water, and processed. All of these scrapings were stored at -20ºC if not processed immediately.
Processing of specimens
Biopsies were homogenized in T.E. (pH 8.0, 0.01M Tris, 0.002 M EDTA) buffer. Smears were prepared on circular slides for counting, and specimens were further processed for extraction of nucleic acids as well as cultured for mycobacteria and other aerobic gram-positive and gram-negative organisms. The smears were stained with Ziehl-Neelsen stain and examined for acid-fast bacilli (AFB) which were counted (19).
Extraction of nucleic acids from biopsies/scrapings. A technique based on the principle of a combined application of earlier-described techniques (1,12) developed during the study was used (15). Briefly, the homogenates/scrapings in T.E. buffer were frozen-thawed and then enzymatically treated at 37ºC first with lysozyme (3 mg/ml) for 2 hr followed by proteinase-k (250 µ g/ml) for 6 hr. To this extract an equal volume of phenol: chloroform (1:1) was added and thoroughly mixed. After centrifugation at 8000 x g x 15 min, the aqueous layer was removed and an equal volume of chloroform: isoamyl alcohol was added. This was again thoroughly mixed and centrifuged again at 8000 x g x 15 min. To the aqueous materials, 2 volumes of chilled ethanol was added and the nucleic acids precipitated. The precipitates were separated by centrifugation at 12,000 x g x 15 min; re-dissolved in lysis buffer (6 M guanidine hydrochloride, EDTA 15 mM, Bmercaptoethanol 1 mM), and rRNA and DNA were fractionated (12).
Extraction of nucleic acids in organisms grown from these specimens. Logphase growths from the mycobacteria (M. scrofulaceum, M. avium, M. avium-intracellulare complex, M. gordonae, M. jiavescens, M. smegmatis, M. fortuitum, M. chelonei, M. phlei) as well as gram-positive (Staphylococcus aureus, S. alhus, Micrococcus, Bacillus cereus, B. subtilis, Corynehacterium xerosis, C. hofmanni) and gram-negative (Escherichia coli, Klebsiella pneumonae, Pseudomonas aerogenosa, Proteus vulgaris, P. mirabilis) organisms isolated from these specimens were harvested. Nucleic acids were isolated by the technique standardized earlier and stored at -20ºC (12).
Gene amplification. The method as described by Williams, et al. (32) and Williams and Gillis (31) was followed for the polymerase chain reaction (PCR).
Primers/probes and conditions for PCR. The primers used for gene amplification (31,32). The have been described earlier The probe used for hybridization was a 212-bp fragment prepared after amplification (31,32). Primers/probes were synthesized in a Pharmacia Gene Assembler Plus by following the procedure detailed in the manual of the instrument and using reagents from Pharmacia, Uppsala, Sweden.
Amplification. Briefly, two 25-mer primers directed the synthesis of a 360-bp fragment, approximately 80% of the 18kDa protein gene of M. leprae. The PCR was performed using recombinant Ampli-Taq polymerase with the Gene Amp Kit (Perkin-Elmer-Cetus Instruments, Norwalk, Connecticut, U.S.A.). Temperature cycling was performed in a programmable Thermal Cycler with three temperature shifts of 94ºC (denaturing, 1 min); 60ºC (primer annealing, 1 min), and 72ºC (extension, 2 min). In all cases, 45 cycles were used for the assay. The amplified product was analyzed by dot-blot analysis and/or by Southern transfer and detection of an amplified fragment of 360 bp by using a 212-bp probe for the intron of the amplified fragment as recommended by developers of this assay (32).
Synthesis and labeling of probes with y p32 ATP. The 212-bp probe was prepared by PCR amplification of an internal region of the 360-bp region of the 18-kDa protein gene using similar conditions and cycle parameters as for the main amplification for the 360-bp fragments. This probe was purified, end labeled with y P32 (using the method described by Manniatis, et al. 18) and was used in the hybridization (31).
Denaturing of amplified DNA and blotting. The procedure as described by Boddinghaus, et al. (1) was followed. The denatured DNA amplified was directly applied on nitrocellulose membrane for dot-blot analysis using the dot-blot apparatus (BRL; Life Technologies, Paisley, Scotland). After blotting, the nitrocellulose membranes were baked at 80ºC for 2 hr in a vacuum oven (Gallenkamp, Loughborough, U.K.).
Southern transfer. The PCR products were electrophoresed with appropriate markers; the gels were observed under ultraviolet light and the bands recorded, and they were then transferred to nitrocellulose membranes (BA85; Schleicher & Schuell GmbH, Dassel, Germany) by the blotting procedure of Southern (29).
Hybridization of PCR product with 212bp probe. The hybridization of the dot-blot filters was carried out as described by Maniatis, et al. (18) with some modifications. After baking the blots, the filters were soaked in 2 x SSC (I x SSC = 0.15 M sodium chloride, 0.015 M sodium citrate), 0.1% (w/v) SDS for 10 min, and then kept in hybridization buffer supplemented with tRNA (10 mg/ml) at 42ºC for 2-4 hr for pre-hybridization. The pre-hybridization buffer (same for hybridization also) containing 5 x Denhardt (0.1% each of ficoll, polyvinyl pyrrolidone (PVP) and bovine serum albumin (BSA) in sterile deionized water] and 5 x SSC was prepared. Ten ml of the pre-hybridization buffer containing 50% of formamide (BDH, Ltd., Poole, U.K.) was added to each polythene bag. After 2-4 hr of incubation, the pre-hybridization buffer was poured off and 0.2 ml of fresh pre-hybridization buffer for each square centimeter of the membrane was added, and the labeled probe 100 µ l (specific activity > 3000 Ci/mmol) was then added. The incubation continued at 42ºC for 16-20 hr with the use of 507r> formamide. When the hybridization process was completed, the filters were taken out of the bags and quickly immersed in a 4 x SSC, 0.1% SDS for 1 hr at 42ºC.
Final washing of the hybridized membranes was done with 2 x SSC containing 0.1% SDS for 1 hr at 50ºC. When washing was completed, the filters were immediately dried and subjected to autoradiography at -70ºC for the desired length of exposure.
RESULTS
Bacteriological status of patients and controls
In the 56 clinically PB (TT/BT/I) leprosy patients' skin, 6 cases had detectable AFB; 53 of the 73 clinically MB (BB/BL/LL) cases were positive for AFB. Various gram-positive (Staphylococcus aureus, S. albus. Micrococcus, Bacillus subtil is, B. cereus, Corynebacterium xerosis, C. hofmanni) and gram-negative (Escherichia coli, Klebsiella pneumonae, Pseudomonas aerogenosa, Proteus vulgaris, P. mirabilis) and mycobacterial strains (M. scrofulaceum, M. avium, M. avium-intracellulare complex, M. gordonae, M. Jlavescens, M. smegmatis, M. phlei, M. fortuitum, M. chelonei) were isolated from these specimens. The details have been published elsewhere (27,28).
Results of gene amplification (PCR)
Sensitivity and specificity. The gene amplification technique used in this study (36) exhibited consistent and reproducible findings. Overall, 41/56 ( 73% ) of clinical PB cases and 68/73 (93%) of MB cases were positive by PCR (Table 1). The single nerve biopsy specimen was also positive by PCR. There were no false-positives in the 50 control biopsies/skin scrapings examined, thus showing the specificity of the technique. There were no false-positive results with DNAs from any of the organisms grown from these specimens and included in this analysis.
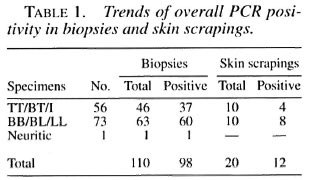
Results of hybridization of PCR product with probe. Overall, a total positivity of 71% (92/130), i.e., single band of appropriate size (360 bp) by ethidium bromide (EB) and blot hybridization was observed. Eighteen cases who had a suspicious band by EB staining were confirmed by probe hybridization and autoradiography. Thus, the overall improvement in the sensitivity was 14% (from 71% to 85%) by hybridization of the PCR product, which is statistically significant (p < 0.005). Further, there were four samples (3%) which were positive for probe hybridization but did not have a visible band on EB staining.
AFB positivity vs positivity with PCR. The data were classified according to AFB positivity/negativity of specimens, and the positivity was analyzed in relation to positive amplification of the 18-kDa gene fragments (Table 2). It was observed that all of the AFB-positive specimens were positive by PCR. Overall, 59/59 (100%) of AFB-positive specimens and 50/70 (71%) of AFB-negative specimens were positive by PCR; 72% (36/50) of smear-negative PB leprosy and 75% (15/20) of smear-negative clinical MB types of cases were positive by PCR.
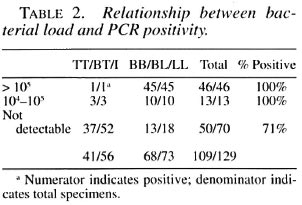
Relationship between load and positivity with PCR. Data were analyzed to assess the relationship between the bacterial load estimated by the AFB counts and PCR (Table 2). The specimens were classified into three groups according to the number of organisms in them. Group I had >105 organisms, Group II had 104 to 105 and Group III had undetectable bacilli. It was observed that PCR positivity in specimens with AFB was 46/46 (100%), 11/11 (100%) and 52/72 (72%), respectively. It is apparent that positivity rates for PCR were significantly higher in specimens with countable organisms. However, there was no difference in PCR positivity in specimens of Group I and Group II (104 and more organisms).
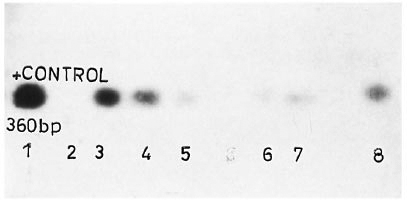
The figure. Positive blot results after Southern transfer and probe hybridization: 1 = Positive control; 2 =negative control; 3, 4 = suspicious on EB staining and positive on blot; 5, 6, 7, 8 = negative by EB staining and pos-itive by blot; others negative by both analyses.
DISCUSSION
Leprosy is considered primarily a disease of the skin and nerves. However, the infection is usually systemic and it manifests as a spectrum from tuberculoid to lepromatous leprosy. The paucibacillary (PB) type of cases, in which it is difficult to demonstrate the bacilli, constitute the major proportion of leprosy in India and several parts of the world. The technical limitations in diagnosing leprosy can be overcome to a great extent by the recent advances in the application of recombinant DNA technology. At present, several probes for various mycobacteria, including M. leprae and M. tuberculosis, have become available. Because of several hypothesized advantages like several thousand copy numbers, many investigators have recently focused on rRNA targeting probes. These rRNA targeting probes (13,15,22) can detect most of the MB cases; however positivity is lower in PB cases. As in other areas of application, the most significant advances in molecular biology of mycobacterial infections has been the development of gene amplification/polymerase chain reaction (PCR). Positive signals from specimens with less than 10 organisms with gene amplification and detection with a labeled probe offer a considerably higher sensitivity than any method known so far. A number of gene amplification techniques targeting various genes of M. leprae have been developed recently and sensitivity ranging from 1-1000 bacilli has been reported (2,4,7,10,20,21,23,31,34,36). PCR techniques have been found useful for a variety of leprosy specimens, including biopsy sections (4). Most of these gene amplification techniques for the detection of M. leprae have been shown to be highly sensitive in MB cases, but have been shown to give variable results in PB cases (4,36). Positivity has ranged from 60%-70% in smear-negative, untreated PB cases in different studies (4,7).
The findings of the present study confirm that the system described by Williams, et al. (36) is appropriate and applicable to patients in this region. The assay exhibited 100% positivity in AFB-positive and 71 % positivity in AFB-negative specimens. Similar observations have been reported by the developers of this assay (7). This substantial positivity in specimens with undetectable AFB will be of immense value in confirmation of the diagnosis in smear-negative, clinically/histologically atypical cases which are emerging as a major problem all over the world. A negative result in nearly 30% of clinically confirmed, smear-negative specimens indicates the need and scope for improvement. The approaches could be better sampling/extraction techniques as well as modifications in the amplification procedure. In the studies on microbial flora from these cases, while there was no difference in the distribution of aerobic gram-positive and gram-negative organisms among different types of leprosy cases and controls (27), members of the MAIS complex were observed to preferentially colonize the skin of lepromatous cases (28). Significantly, the specificity of the system has been confirmed by the absence of any false-positive results with nucleic acids extracted from specimens from controls as well as organisms grown from the specimens from controls and leprosy cases.
During recent years there have been suggestions to restrict the analysis of the PCR product to electrophoresis. The demonstration of fragments of a particular size by EB staining for demonstration of the product has the following theoretical advantages: simple to perform, cost effective and less time consuming. Williams, et al. (31,33) have used the hybridization of the probe to confirm the identity of the PCR product, and have reported that this approach significantly improves the detection, especially in PB specimens (33). In the present study, the positivity could be improved substantially by carrying this analysis up to hybridization with a probe. Not only was the sensitivity improved, the confidence about the specificity also increased since these were graded as suspicious by EB staining (14%). Further, 3% of EB-negative and blot-positive specimens also could be considered positive. Such findings also have been recorded in the use of PCR methods for tuberculosis by Boddinghaus, et al. (1) and de Wit, et al. (5) . Whether the same sensitivity can be achieved by using other strategies, such as the nested primer amplification (23) or by amplification of repetitive gene sequences (10,34,36) or by altering the numbers and other cycle parameters, can be determined only after extensive trials. The negative results in more than one-fourth of the clinically confirmed, smear-negative specimens indicate the need and hope for improvement. The approach could be better sampling/extraction procedures or an alternate approach, such as targeting multiple copy targets. It is suggested that hybridization with a probe should be undertaken for obtaining optimum results.
Acknowledgment. The authors are thankful to LEPRA (U.K.) for the gift of some of the reagents used. Clinical suport by the paramedical staff (S. Thomas, N. Yubana, K. N. Singh, P. John, P. Arora, B. Bhan, D. N. Lal, S. Crispin, S. Haldar, S. Gautam, N. Begum, Ram Murti and N. Crispin); technical support by S. K. Bhan, Noel S. Singh, A. Robi, Shri Ram; photographic assistance by H. O. Agarwal and N. Dubey, and the secretarial assistance of J. D. Kushwah and S. K. Bhan is gratefully acknowledged.
REFERENCES
1. BODDINGHAUS, B., ROGALL, T, FLOHR, T, BLOCKER, H. and BOTTGER, E. C. Detection of mycobacteria by amplification of rRNA. J. Clin. Microbiol. 28(1990)333-346.
2. COX, R. A., KEMPSELL, K., FAIRCLOUGH, L. and COLTSON, M. J. The 16S ribosomal RNA of Mycobacterium leprae contains a unique sequence which can be used for identification by polymerase chain reaction. J. Med. Microbiol. 35(1991)284-290.
3. DATTA, A. K., KATOCH, V. M. and SHARMA, V. D. Effect of pyridine extraction on the acid fastness of mycobacteria. Lepr. India 55(1983)299-304.
4. DE WIT, M. Y. L., FABER, W. R., KREIG, S. R., DOUGLAS, J. T., LUCAS, S., MONTREEWASUVAT, N. R. A., PATTYN, S. R., HUSSAIN, R., PONNINGHAUS, J. M., HARTSKEERL, R. A. and KLATSER, P. R. Application of polymerase chain reaction for the detection of Mycobacterium leprae in skin tissues. J. Clin. Microbiol. 29(1991)906-910.
5. DE WIT, S., STEYN, L., SHOEMAKER, S. and SOGIN, M. Direct detection of Mycobacterium tuberculosis in clinical specimens by DNA amplification, J. Clin. Microbiol. 28(1990)2437-2441.
6. DHARMENDRA and CHATTERJEE, S. N. Examination for Mycobacterium leprae. In: Leprosy Vol. 1. Dharmendra, ed. Bombay: Kothari Medical Publishing House, 1978, pp. 258-262.
7. GILLIS, T. P., WILLIAMS, D. L. and JOB, C. K. Evaluation of PCR for detecting Mycobacterium leprae in treated and untreated leprosy patients. (Abstract) Indian J. Lepr. 64(1992)247.
8. HACKEL, C, HOUARD, S., PORTAELS, F., VAN ELSEN, A., HERZOG, A. and BOLLEN, A. Specific identification of Mycobacterium leprae by the polymerase chain reaction. Mol. Cell Probes 4(1990)205-210.
9. HARTSKEERL, R. A., DE WIT, M. Y. L. and KLATSER, P. R. Polymerase chain reaction for the detection of Mycobacterium leprae. J. Gen. Microbiol. 135(1989)2357-2364.
10. JAMIL, S., WILSON, S. M., HACKET, M., HUSSAIN, R. and STOKER, N. G. A colorimetric PCR method for the detection of M. leprae in skin biopsies from leprosy cases. Int. J. Lepr. 62(1994)512-526.
11. KATOCH, K. Monitoring of chemotherapy in leprosy-a clinician's perspective. Indian J. Lepr. 64(1992)425-427.
12. KATOCH, V. M. and Cox, R. A. Stepwise isolation of RNA and DNA from mycobacteria. Int. J. Lepr. 54(1986)409-415.
13. KATOCH, V. M., KANAUJIA, G. V., SHIVANNAVAR, C. T. KATOCH, K., SHARMA, V. D., PATIL, M. A. and BHARADWAJ, V. P. Progress in developing ribosomal RNA and rRNA gene(s) based probes for diagnosis and epidemiology of infectious disease specially leprosy. In: Tropical Disease-Molecular Biology and Control Strategies. Kumar, S. A., Sen, A. K., Datta, G. P. and Sharma, R. N., eds. New Delhi: CSIR Publication, 1994, pp. 580-587.
14. KATOCH, V. M., KATOCH, K., SHARMA, R. K., SHIVANNAVAR, C. T, SHARMA. V. D., PATILM M. A., BHATIA, A. S. and BHARADWAJ, V. P. Characterization of M. leprae strains by RFLP analysis of amplified rDNA. (Abstract) Int. J. Lepr. 61 Suppl.(1993)114A.
15. KATOCH, V. M., KATOCH, K., SHIVANNAVAR, C. T, KANAUJIA, G. V., SHARMA, R. K., SHARMA, V. D. and PATIL, M. A. Development of improved techniques for extraction of nucleic acids from leprosy lesions. (Abstract) Int. J. Lepr. 61 Suppl.(1993)114A.
16. KHANOLKAR, S. R., MACKENZIE, C. D., LUCAS, S. B., HUSSEN, A., GIRDHAR, B. K., KATOCH, K. and MCADAM, P. W. J. Identification of Mycobacterium leprae antigens in tissues of leprosy patients using monoclonal antibodies. Int. J. Lepr. 57(1989)652-658.
17. LEIKER, D. L. and McDOUGALL, A. C. Technical guide for smear examination for leprosy by direct microscopy. Amsterdam: Leprosy Documentation Service, 1983.
18. MANIATIS, T, FRISTSCH, E. F. and SAMBROOK, J. Molecular Cloning: A Laboratory Manual. Cold Spring Harbor, N.Y.: Cold Spring Harbor Laboratory, 1982.
19 McRAE, D. H. and SHEPARD, C. C. Relationship between the staining quality of Mycobacterium leprae and infeetivity in mice. Infect. Immun. 3(1971)116-120.
20. MISRA, N., RAMESH, V., MISRA, R. S., SHANKARNARAYAN, N. R, COLSTON, M. J. and NATH, I. Clinical utility of LSR/AI5 gene for M. leprae detection in leprosy tissues using polymerase chain reaction. Int. J. Lepr. 63(1995)35-41.
21. PATTYN, S. R., URIS, D., IVEN, M., REES, R. J. W. and JAMES, P. Polymerase chain reaction amplifying DNA coding for species-specific rRNA of Mycobacterium leprae. Int. J. Lepr. 60(1992)234-243.
22. PITULLE, C, WITT, D., STACKEDRANDT, E. and KAZDA, J. Further evidence for the exclusiveness of the Mycobacterium leprae -specific DNA probe. Int. J. Lepr. 58(1990)130-133.
23. PLIKAYTIS, B. B., GELBER, R. H. and SHINNICK T. M. Rapid and sensitive detection of Mycobacterium leprae using nested-primer gene amplification assay. J. Clin. Microbiol. 28(1990)1913-1917.
24. RIDLEY, D. S. Bacterial indices In: Leprosy in Theory and Practice. Cochrane, R. G. and Davey, R. F., eds. London: John Wright and Sons, Ltd., 1964, pp. 620-622.
25. RIDLEY, D. S. and JOPLING, W. H. Classification of leprosy according to immunity: a five-group system. Int. J. Lepr. 34(1966)255-273.
26. SAIKI, R. K., SCHARFS, F. F., MULLIS, K. B., HORN, G. T., ERLICH, H. A. and ARNHEIM, N. Enzymatic amplification of B-globin genomic sequence and restriction site analysis for diagnosis of sickle cell anemia. Science 230(1985)1350-1354.
27. SHARMA, R. K., KATOCH, K., SHARMA, V. D., SHIVANNAVAR, C. T., NATRAJAN, M. and KATOCH, V. M. Isolation and characterization of cultivable mycobacteria from leprosy skin. Indian J. Lepr. 67(1995)309-319.
28. SHARMA, R. K., KATOCH, K., SHARMA, V. D., SHIVANNAVAR, C. T., NATRAJAN, M. and KATOCH, V. M. Studies on microbial aerobic flora of skin in leprosy patients across the spectrum. Indian J. Lepr. 67(1995) 321-328.
29. SOUTHERN, E. M. Detection of specific sequences among DNA fragments separated by gel electrophoresis. J. Mol. Biol. 90(1975)503-517.
30. VENKATESAN, K., MINNIKIN, D. E., SINGH, H., RAMU, G. and BHARADWAJ, V. P. Detection of mycobacterial lipids in skin biopsies from leprosy patients. FEMS Microbiol. Lett. 44(1987)167-172.
31. WILLIAMS, D. L. and GILLIS, T. P. Identification of M. leprae by PCR. In: Proceedings of the Workshop on PCR Technology for the Detection of Mycobacterium leprae, Sasakawa Research Building, Leprosy Division, Thailand, 8-1 9 April, 1991, pp. 18-22.
32. WILLIAMS, D. L., GILLIS, T. P., BOOTH, R. J., LOOKER, D. and WATSON, J. D. The use of a specific DNA probe and polymerase chain reaction for the detection of Mycobacterium leprae. J. Infect. Dis. 162(1990)193-200.
33. WILLIAMS, D. L., GILLIS, T. P., FIALLO, P., Job, C. K., GELBER, R. H., HILL, C. and IZUMI, S. Detection of Mycobacterium leprae and the potential for monitoring anti-leprosy drug therapy directly from the skin biopsies by PCR. Mol. Cell Probes 6(1992)401-410.
34. WOODS, S. A. and COLE, S. T. A rapid method for the detection of potentially viable M. leprae in human biopsies - a novel application of PCR. FEMS Microbiol. Lett. 65(1989)305-310.
35. WHO EXPERT COMMITTEE ON LEPROSY. Sixth report. Geneva: World Health Organization, 1988. Tech. Rep. Ser. 768 .
36. YOON, K. H., CHO, S. N., LEE, M. K., ABALOS, R. M., FAJARDO, T. T, GUIDO, L. S., DELA CRUZ, E. C., WALSH, G. P. and KIM, J. D. Evaluation of polymerase chain reaction-amplification of Mycobacterium leprae specitic repetitive sequence in biopsy specimens from leprosy patients. J. Clin. Microbiol. 31(1993)895-899.
1. Ph.D., Research Associate.
2. M.D., Assistant Director.
3. Ph.D., Research Assistant.
4. Ph.D., Research Officer.
5. M.Stat., Assistant Director.
6. Ph.D., Research Associate.
7. M.D., Deputy Director.
Central JALMA Institute for Leprosy (ICMR), Taj Ganj, Agra 282 001, India.
Reprint requests to Dr. V. M. Katoch.
Received for publication on II December 1995.
Accepted for publication in revised form on 1 May 1996.