- Volume 60 , Number 4
- Page: 549–55
A rapid and sensitive high performance liquid chromatographic analysis of clofazimine in plasma
ABSTRACT
The high performance liquid chromatographic (HPLC) method of Gidoh, et al. has been modified substantially to provide a simple, rapid, and relatively inexpensive procedure for measuring clofazimine in plasma. The modification involves the use of commonly available laboratory reagents instead of custom-made ones. It also employs a solid phase system for efficient extraction instead of the conventional, less efficient and more labor intensive, liquidliquid extraction. The inclusion of an internal standard (salicylic acid) improves the precision and reproducibility. It is demonstrated that the method can be used to monitor in vivo clofazimine levels as may be required in formal pharmacokinetic studies or therapeutic drug monitoring.RÉSUMÉ
La méthode de chromatographic liquide de haute performance de Gidoh, et al. a été modifiée de maniere substantielle de manière à fournir un procédé simple, rapide et relativement peu coûteux de dosage de la clofazimine dans le plasma. La modification concerne l'utilisation de réactifs de laboratoire communément disponibles plutôt que de ceux produits par l'utilisateur. Le procédé emploie aussi un système en phase solide pour une extraction efficiente plutôt que l'extraction liquide-liquide conventionnelle, moins efficiente et plus laborieuse. L'inclusion d'un standard interne (acide salicylique) améliore la précision et la reproductibilité. Il est démontré que la méthode peut être utilisée pour le suivi in vivo des taux de clofazimine comme cela peut être nécessaire pour des études formelles de pharmacocinétique ou la surveillance du traitement médicamenteux.RESUMEN
El método de la cromatografía de líquidos de alta resolución (HPLC) de Gidoh ct al., se ha modificado substancialmente para transformarlo en un procedimiento simple, rápido, y relativamente barato, útil en la medición de la concentración de clofazimina en plasma. La modificación involucra el uso de reactivos comunes en el laboratorio, en lugar de reactivos especiales. También emplea un sistema de extracción en fase sólida que es más eficiente que el convencional procedimiento de extracción liquido-liquido (menos eficiente y más laborioso). La inclusión de un estándar interno (ácido salicilico) mejora la precisión y la reproducibilidad de los resultados. El método puede ser usado para cuantificar in vivo los niveles de clofazimina en estudios farmacocinéticos formales y durante el seguimiento de los pacientes en tratamiento.Clofazimine (CLF) [3-(p-chloranilino)-10(p-chlorophenyl 2,10-dihydro-2-isopropylimino phenazine] is a primary antileprosy drug currently used in the so-called triple combination therapy adopted worldwide by the World Health Organization (11). The drug is also indicated in the treatment of Buruli skin ulcer (9), pyoderma gangrenosum (6), and in the acquired immune deficiency syndrome (AIDS) complicated with Mycobacterium avium complex (MAC) infection (3).
Reported high performance liquid chromatographic (HPLC) methods of analyzing CLF in body fluids are limited. They are either complicated, needing specialized instrumentation and custom-made reagents, or they lack internal standards (5, 10). For instance, the method of Peters, et al. (10) involves laborious and time-consuming liquid-liquid extraction of CLF, employing an HLPC system equipped with a heated (40ºC) analytical column, and it also lacks an internal standard. Gidoh, et al. (5) used specialized (custom-made) commercial reagents of unknown composition and also employed a complicated extraction and column switching procedure.
CLF is used, largely, in the developing countries of Asia, Africa and Latin America where leprosy is endemic. These continents typically also have very limited technical and economic resources. Therefore, a simplified, rapid, and inexpensive procedure to determine CLF levels in patients would be beneficial. Accordingly, we have attempted to simplify the method of Gidoh, et al. (5)by using readily available, common laboratory reagents. Salicylic acid (or benzoic acid) has been used as the internal standard. A solid phase extraction (SPE) procedure has been developed to extract CLF from plasma. The SPE technique is very well accepted now due to its advantages, including high recovery of analyte. Thus, the analytical method is more sensitive and economical with respect to time, solvent, and labor relative to the conventional liquid-liquid extraction.
MATERIALS AND METHODS
CLF was provided by Astra IDL Limited, Bangalore, India. Certified ACS grade salicylic acid was obtained from Fisher Scientific Company, Springfield, New Jersey, U.S.A. The SPE CN column (Prep Sep®) was supplied by Fisher Scientific Company, Ottawa, Canada. All other reagents were of HPLC grade. Water was obtained from an all-glass distillation apparatus. Human plasma from healthy drug-free volunteers was supplied by the Canadian Red Cross Society (Halifax, Nova Scotia), and was used for the preparation of calibration curves and validation of the procedures.
Preparation of standard solutions. A stock solution of CLF was prepared in 50% (v/v) methanol in water and was stored in a refrigerator at 5ºC. Five different standard solutions ranging from 330 µg/l to 6600 µg/l were prepared from the stock solution by diluting with 10% (v/v) methanol in water. A stock solution of salicylic acid (34 Mg/ml) was prepared in 50% (v/v) methanol in water and stored similarly.
Sample preparation and extraction. Plasma samples (1 ml each in five test tubes) were spiked with 100 µl of a standard solution of known CLF concentration. A sixth plasma sample (blank) received only 100 µl of 10% (v/v) methanol in water. The samples were further diluted with 5 ml 0.1 M phosphate buffer (pH 6.0) and mixed with a vortex mixer. The samples were then allowed to stand while the SPE columns were being conditioned.
The SPE columns were mounted on sample preparation manifold (Adsorbex®; E. Merck, Darmstadt, Germany) and condilioned by treating, sequentially, with 3 ml methanol, 5 ml water, and 5 ml 0.1 M phosphate buffer (pH 6.0), taking care not to let the column go dry during the process. Diluted plasma samples were added to the conditioned SPE columns (packed volume of 0.5 ml) and allowed to percolate slowly under the reduced pressure.
After air-drying the column for 2 min, 50 µl of solution containing 170 ng of salicylic acid was added. The analytes were extracted with four 1-ml portions of a solution of tetrahydrofuran (THF), acctonitrile, and methanol in the respective volume ratios of 2:2:1 containing 0.7 mM hexane sulfonic acid (HSA). The eluate was evaporated to dryness in a 37ºC water bath under a stream of nitrogen gas. The residue was reconstituted in 100 µl of mobile phase composed ofTHF, 0.5% acetic acid solution and methanol in the respective volume ratios of 8:11: 1, containing 2.5 mM HSA. Twenty µl of reconstituted sample was injected into the HPLC system using a microsyringe (Hamilton Company, Reno, Nevada, U.S.A.). The peak-height-ratio (PHR) of the standard CLF peak height to the peak of the internal standard (salicylic acid) was measured and plotted against those of the spiked CLF concentrations.
Chromatographic system. The HPLC system comprised a Waters (Milford, Massachusetts, U.S.A.) Model 440 solvent delivery system, ultraviolet detector with a fixed wavelength (280 nm) and a U6K injector system. The analytical column was a 150 × 3.9 mm (i.d.) steel column packed with µ-Bondapak C18 reverse phase particles (Phenomenex, Torrance, California, U.S.A.) fitted with a C18 guard column. Responses were recorded as peaks on a Fisher Recordall Series 5000 recorder (Austin, Texas, U.S.A.) which was operated at a speed of 0.25 cm/min. Mobile phase was clarified and degassed by filtering through a 0.22-µm membrane filter (Millipore Corporation, Bedford, Massachusetts, U.S.A.) under reduced pressure, and was delivered at room temperature at a flow rate of 1.8 ml/min.
Method precision. The precision of the analytical procedure was evaluated by determining the linearity of response over the range of drug concentrations investigated, the detection limit, percentage recovery and reproducibility.
Linearity. A standard calibration curve of PHR versus concentration of spiked standard solution of CLF was assessed by the coefficient of determination obtained by linear regression analysis. The process was repeated on five separate occasions, and the reproducibility was determined by measuring the coefficient of variation of the slopes of the curves.
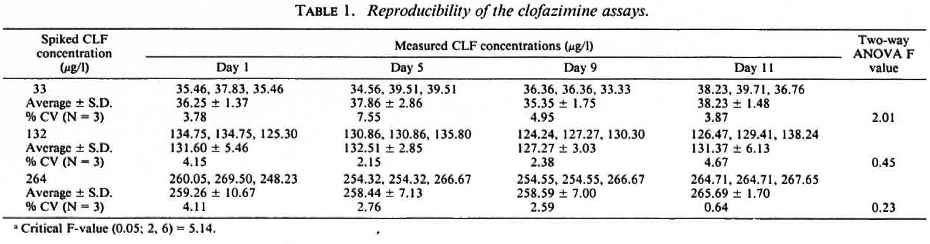
Reproducibility of procedure. The precision of the extraction method was determined by replicate analyses at different times of the day and on different days of plasma samples spiked with three concentrations (33, 132, 264 µg/l) of CLF. Intraday and interday comparisons were made on four separate occasions (days) over a period of 11 days. The mean and coefficient of variation were calculated.
Recovery. The extent of recovery of CLF from the plasma matrix using the SPE procedure was determined by comparing the PHR of the extracted plasma samples containing known amounts of CLF with those obtained by direct injection of the standard solution without treatment.
Application of CLF pharmacokinetics in the rabbit. The analytical procedure was used to study the pharmacokinetics of CFL in a rabbit. The central ear artery of a male. New Zealand, white rabbit (3.5 kg) was cannulated with a Miniset® vein infusion set (Travenol Canada Inc., Ontario, Canada) equipped with a 21-gauge hypodermic needle and winged adapter. This facilitated multiple blood sampling for drug analysis. Patency of the catheter was maintained with intermittent flushing with heparinized (10 U/ml) sodium chloride injection (0.9% NaCl; Abbott Laboratories Ltd., Montreal, Canada). Forty mg of CLF as a 1:10 solid dispersion (7) was packed in a size 0 hard gelatin capsule and administered to the rabbit with sufficient water. Blood samples (3 ml) were removed via the catheter before and at fixed time intervals after drug administration over 24 hr. Blood samples collected in heparinized glass tubes were centrifuged immediately to obtain plasma. The plasma samples were stored at - 20ºC until analysis within 3 days.
On the day of analysis, the plasma samples were allowed to thaw at room temperature and were extracted, along with spiked standard CLF solutions in drug-free rabbit plasma, and analyzed by the HPLC procedure described above.
Data analyses. The calibration curves of the PHR versus spiked CLF concentrations were analyzed by linear regression analysis to obtain the equation of best fit which was then used to calculate the unknown CLF concentrations of the in vivo samples. Interday and intraday variations in the analytical precision were assessed by using the two-way ANOVA regression model with a significance level of p > 0.05.
Concentration-time data following a 40mg oral dose of CLF were fitted to the biexponential equation (Eq. 1) with the aid of the MINSQ computer program (Micro Math Scientific Software, Salt Lake City, Utah, U.S.A.):
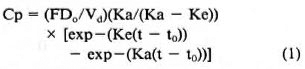
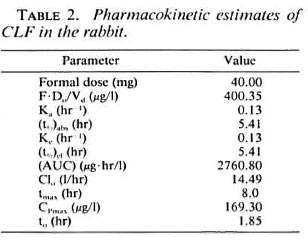
Initial estimates for the curve-fitting were obtained graphically. In Equation 1, Cp was plasma CLF concentration at any time, t, after drug administration, with t0 representing the lag-time prior to drug absorption. F represented the fraction of the formal dose, DQ, which eventually entered the systemic circulation and distributed in an apparent volume, Vd. Ka and Ke were, respectively, the absorption and elimination rate constants which were assumed to be first-order. Secondary parameters including half-lives for absorption (t½)abs and for elimination (t½)el; oral clearance, C1D; apparent initial drug concentration (F-DH/Vd); and total area generated under the concentration-time curve (AUC) were estimated as described by Gibaldi and Perrier (4).
RESULTS
Chromatograms of CLF. The optimal composition of mobile phase used in the procedure for sensitivity and resolution of CLF and the internal standard comprised THF, 0.5% acetic acid solution and methanol in the respective volume ratios of 8:11: 1, containing 2.5 raM HSA. HSA, the ionpairing agent, produced faster elution of CLF, thus reducing its retention time. Typical chromatograms are shown in Figure 1. Figure 1A represents a chromatogram of blank human plasma spiked with the internal standard. No discernible CLF peak was observed within the normal period. Figure 1B represents the chromatogram of human plasma spiked with 264 µg/1 of CLF and 170 ng of the internal standard. The retention times of salicylic acid and CLF were 3.3 and 6.0 min, respectively. Figure 1C is the chromatogram of an in vivo rabbit plasma sample (spiked with the internal standard) 6 hr after oral administration of a 40mg dose of CLF. The peak detected at 6 min is that of CLF. No CLF metabolites were detected.
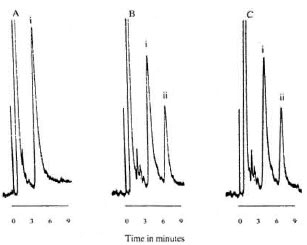
Fig. 1. Chromatograms of salicylic acid (SA, i) and clofazimine (CLF, ii). A = Drug-free human plasma spiked with SA; B = human plasma spiked with CLF and SA; C = rabbit plasma sampled 6 hr after 40-mg CLF (po) and spiked with SA.
Linearity, recovery, detection limit and reproducibility. The PHR obtained with human plasma samples spiked with the concentration range of 16 to 6660 µg/1 were linearly related to the spiked concentrations. The equation of best fit for the line (average of five determinations) was:
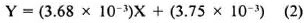
where Y is the PHR and × is the spiked CLF concentration (Fig. 2). The coefficient of determination obtained with the replicate determinations ranged from 98%-100%, and the coefficient of variation (CV) of the slope determined on the five separate occasions was 10.73%.
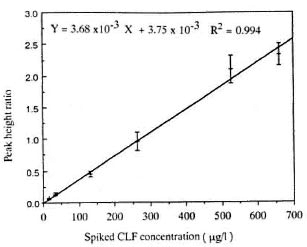
Fig. 2. Calibration curve of PHR versus concentration of spiked standard solutions of CLF. Each point represents the mean ± S.D. of five determinations.
The recovery of CLF from plasma matrix by this procedure was virtually complete (range 100%-108%).
The limit of detection of CLF in plasma was determined by analyzing decreasing concentrations of CLF spiked in drug-free plasma. The minimum detectable concentration when the signal-to-noise ratio was about 3 at 0.005 attenuated unit at full scale (a.u.f.s.) was 3 µg/l.
The reproducibility of determination by the procedure was determined by both intraday and interday variability of the estimates determined at three different times of the day over 11 days. The average % CVs for the intraday variability were 5.04 (range 3.78-7.55), 3.34 (range 2.5-4.57), and 2.53 (range 0.64-4.11), respectively, for the spiked CLF concentrations of 33, 132, and 264 µg/l (Table 1). The interday % CVs (N = 4) for the mean daily CLF concentrations were 3.68, 1.78 and 1.34, respectively. An analysis of variance using the two-way ANOVA regression model showed no significant difference (p > 0.05) between determinations performed on any particular day or from day to day. By implication, the results also indicate that CLF in plasma stored at - 20ºC remained stable over at least 11 days prior to HPLC analysis.
Pharmacokinetics. A plasma CLF concentration-time profile after a single 40-mg dose of CLF as solid dispersion is shown in Figure 3. The pertinent pharmacokinetic parameters estimated are summarized in Table 2.
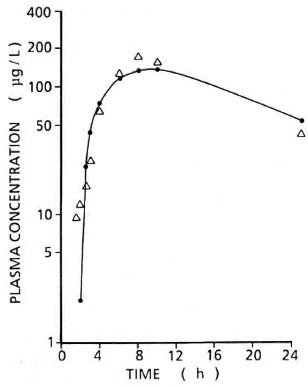
Fig. 3. Log plasma CL F concentration-time profile after a single 40-mg dose (po) in rabbit. ● = Computerfitted concentrations; Δ = experimental concentrations.
DISCUSSION
Linearity of response over a more than 40-fold CLF concentration range has significant advantages in the quantitation in vivo of CLF samples in that such samples need not be diluted to some suitable concentration range prior to analysis. The limit of detection (3 µg/l) in this procedure is comparable to that reported by Lanyi, et al. and Gidoh, et al. (5, 8). Therefore, the method can be used to adequately monitor CLF plasma levels in patients on chronic use of CLF or several weeks after cessation of drug administration.
CLF metabolites were not detected. Feng, et al. (1, 2) estimated that the three metabolites isolated from urine of patients taking CLF on a chronic basis accounted for less than 1% of the administered dose. Since the metabolites have not been shown to be pharmacologically active, they are probably not important from a clinical viewpoint.
Salicylic acid would normally not be used as an internal standard for monitoring drug levels in patients who may also be taking aspirin or other salicylates concomitantly. In the initial attempts to quantitatively monitor CLF plasma levels, benzoic acid (19 ng/ml) was used as the internal standard. It was discovered, however, that in the presence of dapsone (a drug which is commonly coadministered with CLF) the benzoic acid peak was poorly resolved from the solvent front and the dapsone peak. Benzoic acid was, therefore, replaced with salicylic acid to make the method suitable for simultaneous determination of dapsone and CLF plasma levels in a single sample. With salicylic acid as the internal standard, the peaks of salicylic acid, dapsone, and CLF were well separated for quantitation.
As indicated in Figure 4, in the absence of dapsone, or where salicylate consumption is expected, benzoic acid may be used as the internal standard in place of salicylic acid.
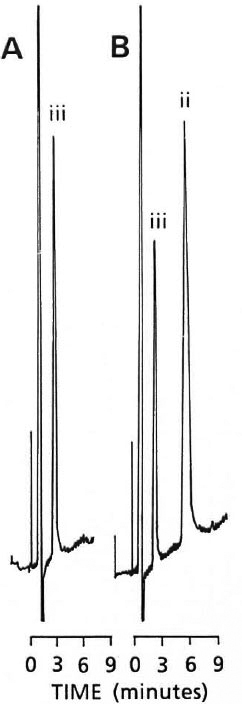
Fig. 4. Chromatograms (0.01 a.u.f.s.) of A = benzoic acid (iii) (25 ng/ml) spiked in drug-free plasma; B = benzoic acid (iii) (19 ng/ml) and CLF (ii) (3.3 µg/ml) spiked in drug-free plasma
Potential interference of the other commonly coadministered antileprosy drug (rifampin) was not investigated.
It is acknowledged that the single-dose pharmacokinetic study was undertaken to demonstrate the applicability of the modified method to the analysis of the pharmacokinetics of CLF in a rabbit, rather than a formal pharmacokinetic study of CLF in the rabbit. Based on the single study, however, one may infer that CLF absorption in the rabbit is slow, delayed, and erratic. The disposition of the single-dose study also indicates that the rates of absorption and elimination proceed at about the sarrie order of magnitude.
Acknowledgment. This study was supported in part by the financial assistance of Novopharm Limited, Ontario, Canada, to which we would like to express our deep appreciation. Thanks are also due to Astra IDL Limited, Bangalore, India, and GA F Chemical Corporation, Wayne, New Jersey, U.S.A., for the free samples of clofazimine and Gantrcz® copolymer, respectively.
REFERENCES
1. FENG, P. C. C, FENSELAU, C. C. and JACOBSON, R. R. Metabolism of clofazimine in leprosy patients. Drug Met. Dispos. 9(1981)521-524.
2. FENG, P. C. C, FENSELAU, C. C. and JACOBSON, R. R. A new urinary metabolite of clofazimine in leprosy patients. Drug. Met. and Dispos. 10(1982)286-288.
3. GARRELTS, J. C. Clofazimine: a review of its use in leprosy and Mycobacterium avium complex in fection. DICP 25(1991)525-531 (54 ref.).
4. GIBALDI, M. and PERRIER, D. One-compartment model. In: Pharmacokinetics. New York and Basel: Marcel Dekker Inc., 1982. pp. 1-43.
5. GIDOH, M. and TsuTSUMI, S. Determination of O. the three main antileprotic drugs and their metabolites in serum by HPLC. J. Chromatogr. 223(1981)379-392.
6. KARK, E. C, DAVIS, B. R. and POMERANZ. J. R. Pyoderma gangrenosum treated with clofazimine. J. Am. Acad. Dermatol. 4(1981)152-159.
7. KRISHNAN, T. R. and ABRAHAM, I. Improved aqueous dissolution of clofazimine from cocvaporates using polyvinylmethyl ether maleic anhy dride copolymer. Drug Develop. Ind. Pharm. 17(1991)1823-1842.
8. LANYI, Z. and DUBOIS, J. P. Determination of clofazimine in human plasma by thin-layer chro matography. J. Chromatogr. 232(1982)219-223.
9. OLUWASANMI, J. O.. SOLANKE, T. F., OLURIN, E. O., ITAVEMI, S. O.. ALABI. G. O. and LUCAS, A. O. Mycobacterium ulcerans (Buruli) skin ulceration in Nigeria. Am. J. Trop. Med. Hyg. 25(1976)122-128.
10. PETERS. J. H., HAMME, K. J. and GORDON, G. R. Determination of clofazimine in plasma by HPLC. J. Chromatogr. 229(1982)503-508.
11. WH O EXPERT COMMITTEE ON LEPROSY. Sixth Report. Geneva: World Health Organization, 1988, pp. 7-51. Tech. Rep. Ser. 768.
1. M.Pharm.; College of Pharmacy, Faculty of Health Professions, Dalhousic University, Halifax B3H 3J5, N.S., Canada.
2. Ph.D.; College of Pharmacy, Faculty of Health Professions, Dalhousic University, Halifax B3H 3J5, N.S., Canada.
Reprint requests to Dr. Abraham.
Received for publication on 22 June 1992.
Accepted for publication in revised form on 8 October 1992.