- Volume 63 , Number 1
- Page: 35–41
Clinical utility of LSR/A15 gene for Mycobacterium leprae detection in leprosy tissues using the polymerase chain reaction
ABSTRACT
Skin biopsy and slit-skin smears F rom 46 leprosy patients and 4 nonleprosy patients were tested for the presence of Mycobacterium leprae by the polymerase chain reaction (PCR) using primers based on the sequence of the LSR/15 kD gene. The PCR was found to be specific and sensitive, with a detection level of 10 and 100 bacilli. PCR using skin biopsies gave a higher detection rate than did slit-skin smears, probably due to the higher density of bacilli in a 4-mm punch biopsy. Dot blot hybridization with radioactive probes was 10-fold more sensitive than the ethidium bromide staining. Eight patients who did not show acid-fast bacilli in tissues by the conventional methods were shown to have PCR-amplificd M. leprae DNA. False-negative results were obtained in 3 cases even though formal evidence for tissue inhibitors was absent.RÉSUMÉ
Une biopsie cutanée et des frottis cutanés de 46 malades de la lèpre et 4 patients non-lépreux ont été testés pour la présence de Mycobacterium leprae par la réaction de polymerase en chaîne (PCR) en utilisant des amorces basées sur la séquence du gène LSR/15kD. On a trouvé la PCR sensible et spécifique, avec un taux de détection de 10 à 100 bacilles. La PCR sur des biopsies cutanées a donné un taux de détection plus élevé que la PCR sur frottis cutanés, probablement du fait de la densité plus élevée de bacilles dans une biopsie en poinçon de 4 mm. Une hybridization par "dot blot" avec des sondes radioactives était dix fois plus sensible que la coloration à l'ethidium-bromide. On a révélé de l'ADN de M. leprae amplifié par PCR pour huit patients qui n'avaient pas montré de bacilles acido-résistants par les méthodes conventionnelles. Des résultats faussement négatifs ont été obtenus dans trois cas malgré l'absence d'évidence formelle d'inhibiteurs tissulaires.RESUMEN
Se usaron biopsias de piel y extendidos de linfa cutánea de 46 pacientes con lepra y de 4 individuos sin lepra, para buscar la presencia de Mycobacterium leprae por la técnica de la reacción en cadena de la polimcrasa (PCR) usando iniciadores basados en la secuencia del gene LSR/15 kD. Se encontró que la PCR fue específica y sensible, con un nivel de detección entre 10 y 100 bacilos. La PCR usando biopsias de piel dio un mayor grado de detección que la PCR con extendidos de linfa cutánea, esto probablemente se debió a la mayor densidad de bacilos en la biopsia de 4 mm. La hibridización con sondas radioactivas fue 10 veces más sensible que la tinción con bromuro de etidio. Ocho pacientes que no mostraron bacilos en sus tej idos por las técnicas convencionales tuvieron DNA de M. leprae amplificado por PCR. En tres casos se observaron resultados falsos negativos aún cuando no hubieron evidencias formales de inhibidores tisulares.In recent years there has been considerable interest in the application of molecular techniques to detect and identify microorganisms in infected tissue. In infections such as leprosy, where the causitivc organism cannot be grown in vitro, and tuberculosis, where the bacillus may take several weeks to culture, such an approach would be particularly attractive. One such technique, the polymerase chain reaction (PCR), has been applied by a number of groups for the detection of Mycobacterium leprae. The PCR has the advantage of being extremely sensitive and potentially highly specific if the sequences which are selected to prime the reaction are themselves specific for M. leprae. A variety of target sequences have been used for the detection of M. leprae. In general, these have cither been DNA sequences which encode major antigens, such as the 18-kDa (15), 36-kDa (3) or 65-kDa (3) proteins, or nonantigen-encoding sequences, such as M. leprae-specific repetitive sequence C 6) or ribosomal RNA sequences 2, 8). In this study, we report the use of another antigen-encoding sequence for the detection of M. leprae in clinical specimens. The sequence we have used is that which encodes the 15-kDA antigen LSR/A15 which has been shown by ourselves and others to be a target for antibody responses in leprosy patients (5,11), particularly those undergoing type 2 reactions (13).
MATERIALS AND METHODS
Patients. The study included tissues from 46 leprosy and 4 nonlcprosy patients attending the Dermatology Department of Safdarjung Hospital, New Delhi, and Voluntary Health Centre, Sakthinagar, Tamil Nadu, India. Leprosy patients were classified by the clinicopathological criteria of Ridley and Jopling (l0). The patients consisted of 1 indeterminate, 9 polar tuberculoid (TT), 14 borderline tuberculoid (BT), 12 polar Icpromatous (LL) and 10 borderline lepromatous (BL) leprosy cases. Two BT and 16 BL/LL patients had received multidrug therapy (MDT) consisting of daily 100 mg dapsone (4,4'-diaminodiphenyl sulfonc), 100 mg of clofazamine [3-(p-chloroanilino)- 10-(p-chlorophcnyle)-2,10-dihydro-2-(isoprapylimino) phenazinc] on alternate days, and 1500 mg of rifampin once a month for varying periods. One patient each with tuberous sclerosis, toxic melanoma, lupus vulgaris and discoid erythematous plaques were included as negative controls. The bacterial index (BI) was scored on a logarithmic scale by slit-smear examination for acid-fast bacilli (AFB) obtained from six sites (9). Two 4-mm punch biopsies were taken from all patients. One biopsy was used for histological confirmation of diagnosis and the other for DNA extraction.
Mycobacterial isolates and purification of mycobacterial DNA. DNA was prepared from a) armadillo-derived purified M. leprae obtained from Dr. R. J. W. Rees, National Institute for Medical Research, Mill Hill, London, U.K., through the IMMLEP program of the World Health Organization; b) skin biopsies from leprosy and nonleprosy patients and c) slit-smear tissue extracts from leprosy patients using essentially similar methodologies. DNA from other mycobacterial strains were kindly provided by Dr. J. S. Tyagi, All India Institute of Medical Sciences, New Delhi (1).
Leprosy bacilli were obtained from human biopsies as previously described (15). In brief, the tissues were minced with scissors, followed by homogenization in 2 ml of RPMI 1640 (GIBCO BRL, Irvine, Scotland) using glass homogenizers (Wheaton Scientific, Millvillc, New Jersey, U.S.A.). Tissue debris was allowed to settle at 300 g for 5 min and the supernatant containing AFB collected. Nonleprosy biopsies obtained from other dermal lesions were treated in a similar manner. Slit-smear extracts from the carlobes and lesions of leprosy patients were obtained with sterile scalpels which were then washed in 1 ml of sterile deionized water in 1.5-ml vials. The AFB were counted (12) and the samples stored at -70ºC until further use.
M. leprae in biopsy samples and slit smears were pelleted and resuspended in 50 ml of sterile water. Cells were then lyscd by initially boiling for 5 min followed by five consecutive freeze-boil cycles lasting for 1 min each.
Polymerase chain reaction (PCR). PCR was performed essentially as described earlier (7). The primers were selected on the basis of the nucleotide sequence of the LSR/ A15 gene encoding the 15-kDa antigen of M. leprae (5,11) from two homologous regions separated by a DNA segment of 321 bp. The oligonucleotides used were 290-309 bp CGAAGAAGTGACCGTCAACC and 609-628 bp AATGCGTCAAT-GACGTCGGC.
Aliquots of 10 µl of M. leprae DNA, human DNA and DNA of other bacterial and mycobacterial species were added to 5 µl of 10 x PCR buffer (Northumbria Biologicals, Cramlington, U.K.); 8 jd of dcoxynuclcotidc triphosphate (dNTP, 1.25 mM; Pharmacia Chemicals, Upsala, Sweden); 0.25 µl Taq polymerase (200 U/µl, NBL), and 2.5 µl each of 20 mM primers in a 0.5-ml microfuge vial; 100 µl of mineral oil (Sigma Chemical Co., St. Louis, Missouri, U.S.A.) was overlaid on each sample. The reaction mixture was subjected to 35 cycles of 1 min each at 95ºC, 48ºC, and 72ºC followed by a final extension at 72ºC for 5 min in a thcrmocyclcr (Cambio 1HB 2024, Cambridge, U.K.).
The positive controls consisted of DNA from 0.1 -10 ng of the recombinant plasmid pYA1292 with the complete LSR gene kindly provided by Dr. J. Clark-Curtiss, Washington University, St. Louis, Missouri, U.S.A. (11). Negative controls consisted of reaction mixtures in the absence of DNA. The samples were used neat and at a 1:10 dilution to control for inhibitors in the tissue extracts. Negative samples were re-tested with an internal control consisting of known M. leprae DNA subjected to PCR amplification as above with the aim of detecting inhibitors in the test samples.
PCR products were routinely visualized by applying 10µl samples on 2% agarose gels and staining with cthidium bromide after the run (6).
Dot blot hybridization. Dot blot hybridization was undertaken essentially to check the amplified product for sensitivity and specificity using the 32P-labclcd A15 probe. DNA samples of 50 µl were denatured by the addition of 280 µl of 3 N NaOH, heating for 30 min at 72ºC, and blotted using dot blot apparatus (Schleicher and Schucll, Dassel, Germany). The blot was then dried, baked and used for hybridization with the radiolabeled probe. Hybridization signals were detected by autoradiography (6).
RESULTS
Preliminary studies were undertaken to check the specificity and sensitivity of the PCR by using the M. leprae (LSR/A15) gene along with preparations of well-characterized bacteria (including both mycobacterial and non-mycobactcrial species) and purified M. leprae.
PCR specificity. Both primers were tested in the PCR using target DNA purified from 24 bacterial and mycobacterial strains. Specific amplification was observed only with M. leprae DNA (Fig. 1). Of particular importance was the absence of amplified product in the DNA of mycobacterial species that are closely related to the leprosy bacillus or act as causative dermal pathology such as M. tuberculosis. M. scrofulaceum and M. avium-intracellularc group. The size of the amplified fragment was 321 bp, which is in agreement with the position of the primers in the LSR/ A15 gene (5,11).
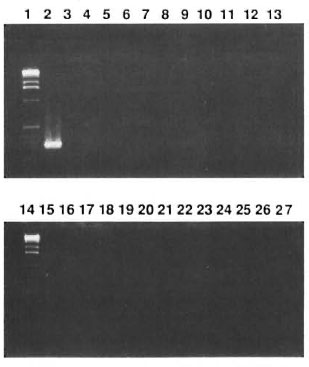
Fig. 1. Specificity of 321-bp fragment of LSR/A15 gene of M. leprae as checked on agarose gel and stained with ethidium bromide. Positive signals were seen in Lane 2 with M. leprae but not in Lanes 3 to 26 which had, in sequential order, DNA from M. tuberculosis [3], M. boris [4], M. bovis (BCG)[5], M. vaccae [6], M.smegmatis [7], M. scronfoncelus [8] M. gastri [9] M.gordonae[10] M. kansasii [11], M. fortuimtum [12] M.cheloni [13], M. intracellulare[15], M. alricanum [16],M. phlei [17], M. avium [18], M. Microti [19], human PBMC [20], P. aureginosa [21], S. pneumoniae [22], S. aureus [23], K. pneumoniae [24], E. coli [25], C.diphtheriae [26]. Lanes 1 and 14 contain a -kb DNA ladder as the molecular weight marker. Lane 27 contains DNA-free control.
PCR sensitivity. The sensitivity of the PCR was determined by testing serially diluted samples of a stock DNA extracted from 106 armadillo- and human-derived leprosy bacilli. Reproducible and unequivocal bands were detectable up to a lower limit which was equivalent to 100 bacilli in ethidium bromide stained agarose gels (Fig. 2A). Sensitivity was improved by dot blot hybridization such that DNA from the equivalent of 10 bacilli gave detectable signals (Fig. 2B).
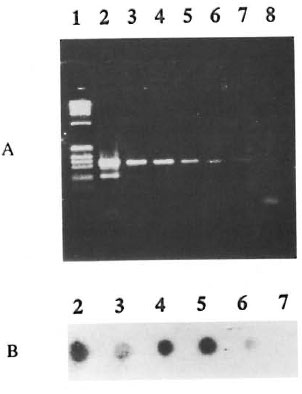
Fig. 2. Sensitivity of 321-bp PCR fragments ofl-SR/Al5 gene of M. leprae as visualized by (A) ethidium bromide staining of electrophoresed agarose gel and (B) autoradiography of dot blot hybridization using 32P-labeled Al5 gene probe. Numbers of M. leprae persample were 105, 104, 103, 102, 10 and 1 in lanes/dots 2, 3, 4, 5, 6 and 7, respectively. Lanes I and 8 containa 1-kb DNA ladder and negative control, respectively.
Detection of M. leprae in human tissues. Eighty-seven infected tissue samples from 46 leprosy and 4 nonlcprosy patients were included in the study. In order to make the PCR amenable for routine clinical testing, a simplified protocol and nonradioactive detection system was employed. Thus, the homogenatcs of skin and slit-smear material were used directly without any purification of bacilli. Negative results were further checked for tissue inhibitors by diluting the sample 1:10 and by incorporating an internal control consisting of known amplifiablc M. leprae DNA in the biopsy and smear extracts. Human DNA from skin material did not have an appreciable inhibitory effect on the PCR. Ethidium bromide staining rather than dot blot hybridization was undertaken for routine screening. Skin biopsies and slit smears of the same individual were tested concomitantly. In addition, conventional BI scores from six sites and AFB counts from extracted material were compared with the PCR results.
Of the nine polar tuberculoid patients tested only one showed a distinct 321-bp amplicon. This correlated well with the presence of AFB in the smear (Table 1). As expected, the PCR positivity increased toward the lepromatous pole. Two of the 14 BT patients gave positive PCR signals with DNA from skin biopsies and one from the slit smears. Of these, only one had microscopically detectable bacilli.

The lepromatous group consisted of freshly diagnosed as well as treated patients with varying quantities of AFB in their tissues as judged by conventional staining methods. In general, there was good correlation between the conventional and PCR methodologies (Table 2). Of interest was the additional detection of bacillary DNA by PCR in two BL and three LL patients who had shown no evidence of AFB. PCR failed to detect M. leprae DNA in slit-smear extracts of one BL and two partially treated lepromatous patients. The reason for this could not be fully attributed to the presence of inhibitors in the tissues since the 1:10 diluted extracts were also negative and the samples spiked with M. leprae DNA showed no decrease in the amplified signal.
Thus, there was concordance for PCR and conventional staining methods in 37 slit smears and 37 skin biopsies for both negative and positive evidence of bacilli. In two slit smears and seven biopsies PCR was a more sensitive method for detecting the presence of M. leprae. Furthermore, positivity was not observed in the four nonleprosy skin lesions.
DISCUSSION
In this study, using the LSR/A15 gene as the target sequence, we have developed a PCR approach for detecting M. leprae which is both sensitive and specific. LSR/A15 is a single copy gene which encodes a protein of unknown function. Primers based on this sequence gave a PCR product in the presence of M. leprae DNA but not when DNA from a variety of other bacteria, including closely related mycobacterial species, was used. Using ethidium bromide stained gels we were able to detect the equivalent of 100 bacilli; this sensitivity was increased a further tenfold when dot blot hybridization using a radio-labeled probe was used.
The detection for PCR-amplified M. leprae DNA was demonstrable across the leprosy spectrum both in 4-mm skin biopsies as well as in slit smear extracts, although the biopsies were more satisfactory presumably because of the larger numbers of bacilli in the 4-mm biopsy samples. In 40 patients both conventional and PCR data were available for both types of tissue samples. Detection of M. leprae by the PCR method was more sensitive in 8 patients; of these, 2 belonged to the paucibacillary TT/BT group and 3 were fully treated lepromatous patients with Bl levels of O. These results emphasize the potential of the PCR for detecting small numbers of bacilli which are undetectable by conventional techniques.
PCR tests for the presence of M. leprae based on a variety of DNA sequences have been reported (2,3,8,15,16). The technique also has been applied to the direct detection of rifampin-resistant M. leprae and M. tuberculosis (4-14). Thus, PCR could become a useful additional tool in clinical mycobacterial laboratories, at least for a small proportion of specimens where conventional techniques are inadequate, and in research laboratories for confirming the identity of putative M. leprae cultures. The current study provides an additional parameter for the identification of the leprosy bacillus.
Acknowledgment. Namita Misra is a recipient of the Senior Research Fellows-hip of the Council for Scientific and Industrial Research (CSIR), India. This study was supported by the Commission of the European Communities, Contract C11*-CT91-0087.
REFERENCES
1. BHARGAVA, S., TYAGI, A. K. and TYAGI, J. S. tRNA genes in mycobacteria: organization and molecular cloning. J. Bacteriol. 172(1990)2930-2934.
2. COX, R. A., KEMPSELL, K., FAIRCLOUGH, L. and COLSTON, M. J. The 16S ribosomal RNA of Mycobacterium leprae contains a unique sequence which can be used for identification by the polymerase chain reaction. J. Med. Microbiol. 35(1991)284-290.
3. HARTSKEERL, R. A., DE WIT, M. Y. L. and KLASTER, R. R. Polymerase chain reaction for the detection of Mycobacterium leprae. J. Gen. Microbiol. 135(1989)2357-2364.
4. HONORE, N., PERRANI, E., TELENTI, A., GROSSET, J. and COLE, S. T. A simple and rapid technique for the detection of rifampin resistance in Mycobacterium leprae. Int. J. Lepr. 61(1993)600-604.
5. LAAL, S., SHARMA, Y. D., PRASAD, H. K., MURTAZA, A., SINGH, S., TANGRI, S., MISRA, R. S. and NATU, I. Recombinant fusion protein identified by lepromatous sera mimics native M. Leprae in T-cell responses across the leprosy spectrum. Proc. Natl. Acad. Sci. U.S.A. 88(1991)1054-1058.
6. MANIATIS, T., FRITCH, E. F. and SAMBROOK, J. Molecular Cloning; a Laboratory Manual. Cold Spring Harbor, New York: Cold Spring Harbor Laboratory, 1982, pp. 150-172 and 470-472.
7. MULLIS, K. B. and FALOONA, F. A. Specific synthesis of DNA in vitro via a polymerase catalyzed chain reaction. Methods Enzymol. 155(1987)335-350.
8. PATTYN, S. R., URSI, D., IEVEN, M., RAES, V. and JAMET, P. Polymerase chain reaction amplifying DNA coding for species-specific rRNA of Mycobacterium leprae. Int. J. Lepr. 60(1992)234-243.
9. RIDLEY, D. S. Bacterial indices. In: Leprosy in Theory and Practice. Cochrane, R. G. and Davey, T. F., eds. Bristol: John Wright and Sons Ltd., 1994, pp. 620-622.
10. RIDLEY, D. S. and JOPLING, W. H. Classification of leprosy according to immunity; a five-group system. Int. J. Lepr. 35(1966)255-273.
11. SELA, S., THOLE, J. E. R., OTTENHOFF, T. H. M. and CLARK-CURTISS, J. E. Identification of Mycobacterium leprae antigens from a cosmid library: characterization of a 15-kilodalton antigen that is recognized by both the humoral and cellular immune systems in leprosy patients. Infect. Immun. 59(1991)4117-4124.
12. SHEPARD, C. C. The experimental disease that follows the injection of human leprosy bacilli into foot pads of mice. J. Exp. Med. 112(1960)445-454.
13. SINGH, S., SHANKERNARAYAN, N. P., JENNER, P. J., RAMU, G., COLSTON, M. J., PRASAD, H. K. and NATH, I. Sera of leprosy patients with type 2 reactions recognize selective sequences in Mycobacterium leprae recombinant LSR protein. Infect. Immun. 62(1994)86-90.
14. TELENTI, A., IMBODEN, D., MARCHESI, F., LOWRIE, D., COLE, S., COLSTON, M. J., MATTER, L., SCHOPFER, K. and BODMER, T. Detection of rifampin-rcsistancc mutations in Mycobacterium tuberculosis. Lancet 341(1993)647-650.
15. WILLIAMS, D. L., GILLIS, T. P., BOOTH, R. J., LOOKER, D. and WATSON, J. D. The use of specific DNA probe and polymerase chain reaction for the detection of Mycobacterium leprae. J. Infect. Dis. 162(1990)193-200.
16. WOODS, S. A. and COLE, S. T. A rapid method for the detection of potentially viable Mycobacterium leprae in human biopsies: a novel application of PCR. FEMS Microbiol. Lett. 65(1989)305-310.
1. M.Sc, Senior Research Fellow;. Department of Biotechnology, All India Institute of Medical Sciences, New Delhi, India.
2. M.D., F.R.C.Path., Department of Biotechnology, All India Institute of Medical Sciences, New Delhi, India.
3. M.D., Consultant; Department of Biotechnology, All India Institute of Medical Sciences, New Delhi, India.
4. M.D., Head, Department of Dermatology, Safdarjung Hospital, New Delhi 1 10029, India.
5. M.B.B.S., D.T.M.N.H., VHS Leprosy Project, Sakthinagar, Tamil Nadu, India.
6. Ph.D., Division of Mycobacterial Diseases, National Institute for Medical Research, Mill Hill, London NW7 1AA, U.K.
Reprint requests to Dr. Indira Nath, Professor and Head, Department of Biotechnology, AIIMS, New Delhi, India.
Received for publication on 25 July 1994.
Accepted for publication on 30 August 1994.