- Volume 62 , Number 4
- Page: 512–20
A colorimetric PCR method for the detection of M. leprae in skin biopsies f rom leprosy patients
ABSTRACT
A one-tube nested polymerase chain reaction (PCR) method for the diagnosis of paucibacillary leprosy was developed using the repetitive RLEP sequence as a target. Detection of the PCR products was simplified by the adaptation of a colorimetric method. The test was specific for Mycobacterium leprae, and the sensitivity of the assay was 1 fg of purified genomic M. leprae DNA (less than one genome). Complete concordance was seen between the development of color and resolution on agarose gels. The results of frozen skin sections f rom untreated patients showed that the assay could detect 100% of multibacillary samples [bacterial index (BI) of 2 or more] and 69% and 70% of the samples with Bis of 1 and 0, respectively. The use of one-tube nested PCR in assessing the effectiveness of multidrug therapy (MDT) in leprosy also was determined. The simplified colorimetric assay was found to be sensitive, rapid and specific, and is suitable for use in routing diagnostic laboratories.RÉSUMÉ
Une méthode de réaction de polymerase en chainc (PCR) en un tube pour le diagnostic de la lèpre paucibacillairc a été développée en utilisant la séquence répétitive RLEP comme cible. La détection des produits de la PCR a été simplifiée par l'adaptation d'une méthode colorimétriquc. Le test était spécifique pour Mycobacterium leprae, et la sensibilité du test était de 1 fg d'ADN génomique purifié de M. leprae (moins d'un génome). Une concordance complete a été observée entre le développement de couleur et la résolution en gels d'agarosc. Les résultats des coupes cutanées réfrigérées provenant de malades non traités ont montré que le test pouvait détecter 100% des échantillons multibacillaircs [ indice bactérien (IB) de 2 ou plus ] et 69% et 70% respectivement des échantillons avec un IB de 1 et 0. L'utilité de la PCR en un tube a également été déterminée pour évaluer l'elficacité de la polychimiothérapie (PCT) de la lèpre. Le test colorimétriquc simplifié a été trouvé sensible, rapide et spécifique, et convient à l'emploi dans des laboratories de diagnostic de routine.RESUMEN
Se usó la técnica de la reacción en cadena de la polimcrasa (PCR) para establecer cl diagnóstico de la lepra paucibacilar utilizando como blanco la secuencia repetitiva RLEP. La detección de los productos amplificados por PCR se simplificó utilizando un método colorimétrico. La prueba fue específica para Mycobacterium leprae, y la sensibilidad del ensayo fue de 1 fg de DNA genómico purificado de M. leprae (menos de un genoma). Se observó una completa concordancia entre el método colorimétrico y la resolución en geles de agarosa. Los resultados de la técnica PCR utilizando secciones congeladas de piel de pacientes con lepra no tratada, mostraron que el ensayo pudo detectar el 100% de las muestras multibacilarcs (índice bacteriano, IB, de 2 o más), y el 69% y 70% de las muestras con IBs de 1 y 0, respectivamente. También se determinó la utilidad de la técnica PCR para establecer la efectividad de la poliquimioterapia en la lepra. El ensayo colorimétrico simplificado fue sensible, rápido y específico; es, por lo tanto, adecuado para su uso rutinario en los laboratorios de diagnóstico.Mycobacterium leprae, the etiologic agent of leprosy, remains one of the few pathogens that cannot be cultivated in vitro. The diagnosis of leprosy is, therefore, still based upon principles used a century ago: clinical examination of the patient's lesions, demonstration of acid-fast bacilli (AFB) in slitskin smears, and histopathology. Although in the majority of cases the diagnosis is clear cut, in early disease or in the indeterminate forms of leprosy clinical evidence may be inconclusive and AFB difficult to find. Furthermore, AFB can be observed only if they arc present in a concentration of approximately 104/ml (14). For the diagnosis of early disease, initiation of effective treatment and monitoring response to chemotherapy, there is a need to develop a method that could specifically detect small numbers of AFB (<104/ml).
Owing to its remarkable sensitivity, the polymerase chain reaction (PCR) has an immense potential in the diagnosis of infectious diseases, particularly where the causative agents are difficult to culture. PCR tests for the diagnosis of leprosy have been developed where single pairs of primers amplify sequences within the genes encoding 18-kDa (17), 36-kDa (pra) (8), 65-kDa (7,20) antigens and rRNA (17). In addition Woods and Cole (20) have developed a test based on a repetitive sequence (RLEP) which is repeated 28 times in the M. leprae chromosome. These tests can detect between 10 to 100 bacilli and arc far more sensitive than any other available method.
The sensitivity and specificity of PCR can be improved further by an additional reaction which follows the first round of amplification. This approach utilizes primers which hybridize to sequences internal to the original primers (12). The nested strategy has an advantage because of the addition of the fresh reagents, the greater number of cycles, and dilution of amplified products reduce the inhibitory factors present. However, it involves extra manipulations, is more expensive, and more prone to contamination. One way of getting around some of these problems is by using a one-tube nested (OTN) PCR (10) which circumvents the requirement for opening the tubes to add the fresh reagents and thereby reduces the risks of contamination; moreover there is only one reaction so it involves fewer manipulations and is more cost-effective.
The use of PCR in a routine diagnostic setting depends upon its speed, sensitivity, reliability and ability to screen large numbers of samples. However, the current method of detection of PCR products is unsuitable for large-scale screening. Different methods have been developed which have simplified the detection and have thus enhanced the utility of PCR in routine diagnostic applications. These methods rely upon the incorporation of modified primers into PCR products to readily allow immobilization and subsequent detection of the amplified products by the development of color. One such colorimetric method has already been developed in conjunction with OTN PCR for the detection of M. tuberculosis (19).
In this report we describe the development of OTN PCR based on the detection of a target within the RLEP sequence of M. leprae. The repetitive nature of this target gives it an advantage in terms of sensitivity over systems detecting single-copy targets. Furthermore, the method of detection of amplified products was simplified to enable large-scale screening of the samples. The usefulness of OTN PCR in the detection of small numbers of M. leprae in tissue samples was determined also. In addition, the method was made quantitative by the use of limiting dilutions and applied to monitoring of the efficacy of multidrug therapy (MDT) in leprosy.
MATERIALS AND METHODS
Patients' samples.
Punch biopsies (4 mm) were collected from untreated leprosy patients presenting at the Marie Adelaide Leprosy Centre, Karachi. The biopsies were snap-frozen in liquid nitrogen and stored frozen until use. Patients were classified according to the Ridley and Jopling scale (13). Punch biopsies were also obtained from 7 LL/BL patients and 1 indeterminate patient receiving MDT, prior to treatment and at 3-, 6-, 12-, and 24-month intervals after the start of therapy. The nonendemic samples consisted of frozen skin biopsies from 8 patients with a variety of other nonleprosy disorders receiving treatment at University College Hospital, London.
M. leprae DNA preparation for PCR. Genomic M. leprae DNA was kindly provided by Dr. M. J. Colston (NIMR, London). The M. leprae DNA was diluted in sterile distilled water to achieve concentrations from 1 ng to 100 ag/2.5 µ l.
Cryostat AS 600 (Anglia Scientific, Cambridge, U.K.) was used to cut 5- µ m sections of each biopsy. The cryostat was cleaned thoroughly between each sample. Individual frozen sections were resuspended in PCR buffer containing 0.5% Tween-20 and 100 µ g/ml proteinase-K (Boehringer Mannheim, Lewes, U.K.) and incubated at 60ºC for 1 hr. This was followed by 10 consecutive freeze boil cycles (20). Fifty yu l of chloroform were emulsified with each sample and then the tubes were centrifuged for 2 min at 13,000 rpm. A 5- µ l aliquot from the aqueous layer was used in a 25- µ l amplification reaction.
PCR primers. Primers PS3 and PS4 hybridize to a specific sequence within the RLEP sequence of M. leprae and generate a 455-bp fragment. Primers NO3 and NO4 bind specifically to sequences internal to the outer primers and generate a 320-bp fragment. Both of these primer pairs were designed using the PRIMER software (Version 0.5; Lincoln, S. E., Daly, M. J., and Lander, E. S.; Whitehead Institute for Biomedical Research, 1991) and synthesized using phosphoramidite chemistry on a Gene Assembler Plus (Pharmacia Biotech Ltd., Milton Keynes, U.K.). Sequences for primers are: PS3, 5'-GGA CAC GAT TAG CGC GGC GCA CGT; PS4, 5'-TTG TGG TGG GCT GGT GGG GTG TGG; NO3, 5'-TGC TGA AGG CGA TAT; NO4, 5'-AGG TTG CCG TAT GTG. Primers NO3 and NO4 were labeled at their 5' end with digoxigenin (British Biotechnology Ltd., Abingdon, U.K.) and biotin, respectively.
Hot start PCR. Hot start was accomplished by overlaying a mixture of all four primers in a reaction tube with a wax pellet. The wax was melted by incubation at 80ºC for 10 min. After a brief centrifugation, the wax was allowed to solidify at room temperature to form a physical barrier above the primers. The other reaction components were layered on top of the wax and only mixed with the primers during the first denaturation step when the wax melted and the two layers were able to mix.
PCR. A 5- µ 1 aliquot of each sample was assayed in a 25- µ l reaction containing 10 mM Tris-HCl pH 8.8; 50 mM KC1; 1.5 mM MgCl2; 0.1% Triton X-100; 200 µ M each of dATP, dCTP, dGTP, and dUTP, 4% of dimethylsulfoxide; 0.75 nmol of each of the outer primers; 12.6 nmol of each of the inner primers, and 0.75 units of Taq polymerase (Bioline, London, U.K.). All of the reactions were carried out on a Hybaid TR2 thermal cycler with the temperature monitored using a thermocouple in a dummy tube.
OTN PCR. For OTN PCR the cycler was programmed to amplify DNA in two different stages. The first stage consisted of 30 cycles of annealing at 68ºC for 30 sec, during which a 455-bp fragment was generated by the outer primers. During the second stage, 20 cycles of annealing at 50ºC for 30 sec were performed, resulting in the amplification of a 320-bp nested fragment by the two inner primers. An initial denaturation temperature of 94ºC for 3 min and subsequent denaturation and extension temperatures of 93ºC for 30 sec and 72ºC for 90 sec, respectively, were used throughout.
Specificity of the primers. The specificity of both the primer pairs for M. leprae DNA was tested separately. DNA was extracted from 18 different mycobacterial species: M. asiaticum, M. avium, M. bovis, M. chelonei, M. duvalli, M.flavescens, M.fortuitum (peregrinum, ranae), M. gordonae, M. marinum, M. nonchromogenicum, M. phlei, M. scrofulaceum, M. smegmatis, M. terrae, M. tuberculosis, M. ulcerans, M. vaccae, and M. xenopi. The outer primer pair of PS3 and PS4 was tested at two different concentrations, 4 nmol and 0.75 nmol; the amplification program consisted of denaturation at 94ºC for 3 min and 50 cycles of 30 sec at 93ºC, 30 sec at 68ºC, and 90 sec at 72ºC, with a final incubation at 72ºC for 10 min. In a separate experiment, primers were tested with the OTN PCR conditions described previously but without the inner primers. The specificity of the inner set of primers was similarly tested for 50 cycles at an annealing temperature of 50ºC and in a mock OTN PCR without the outer primers.
Inhibition controls. The presence of inhibitors in the clinical samples was determined by a method described previously (9).
Quantitation of PCR by limiting dilution. Samples from patients being treated with MDT were first diluted 1/5 in sterile distilled water and then serial twofold dilutions of the samples were made. The highest dilution showing amplification was taken as the PCR titer.
Colorimetric detection of PCR product. The method for colorimetric detection described by Wilson, et al. (19) was adapted for the detection of M. leprae. Microtiter plate wells were coated with 10 µ g/ml of avidin (Sigma Chemical Co., Poole, U.K.) in 50 mM carbonate buffer (pH 9.6) for 1 hr at 37ºC. The reaction tubes were heated to 80ºC to allow the wax to melt, 200 µ l of Tris-buffered saline pH 7.5 with 0.5% Tween-20 (TBS-Tween 20) was added to each tube. After washing the microtiter plates twice with TBS-Tween 20, 100 µ l of each diluted product was added to the individual wells in duplicate, and the plates were incubated for 30 min at room temperature. The plates were washed twice with TBS-Tween 20 and then 100 µ l of 1/5000 of anti-digoxigenin-alkaline phosphatase conjugate (Boehringer-Mannheim) in TBS-Tween 20 was added. After a 30-min incubation at room temperature the plates were washed four times with TBS-Tween 20 and once with 50 mM carbonate buffer pH 9.6; 100 µ l of 1 mg/ml para-nitrophenol substrate (Sigma) in 50 mM carbonate buffer pH 9.6 and 1 mM of MgCl2 was added, and the optical density (OD) was read after 1 hr at 405 nm.
RESULTS
OTN PCR. In order to obtain the greatest sensitivity, the most suitable target for PCR in M. leprae is the RLEP sequence, which is present in 28 copies in the genome (21) and which has previously been used by Woods and Cole (20). When we initially rechecked the specificity of the primers (C and D) described in that study, we found crossreactivity in the DNA from other mycobacterial species. In some cases bands of a size different from M. leprae were seen, but bands of a similar size were observed with DNA from M. fortuitum-ranae and M. ulcerans. This suggests both nonspecific amplification and the presence of elements related to the RLEP sequence in some species. Wc, therefore, designed new primers for an OTN PCR: 24-bp outer primers (PS3/PS4) and 15-bp inner primers (NO3/NO4). The positions of the different primer pairs are shown in Figure 1. PS3/PS4 amplified a 455bp fragment of M. leprae DNA and 320-bp of this sequence was amplified by NO3 / NO4. The specificity of each primer pair alone was tested rigorously, using 1 ng of genomic DNA from different species and 50 cycles of PCR. Initial experiments showed some nonspecific amplification, but the use of a hot-start procedure (1) greatly improved the results (data not shown). All subsequent experiments therefore were carried out using hot-start. This also helped prevent the formation of primer dimers, which is crucial in the use of OTN PCR. With the outer primer pair of PS3 and PS4, no amplification was observed with DNA from 18 mycobacterial species other than M. leprae. However when the annealing temperature (initially 68ºC) was dropped to 50ºC after 30 cycles as would occur in OTN PCR, M. chelonei DNA showed a faint band of high molecular weight. With the internal primer pair NO3 and NO4, after 50 cycles at an annealing temperature of 50ºC, nonspecific bands were seen with several species. In addition, M. avium, M.flavescens, M. fortuitum-peregrinum, and M. tuberculosis (one of the three clinical isolates) showed amplification of a fragment of approximately 320 bp (Fig. 2a). However, using OTN PCR conditions (30 cycles of 68ºC followed by 20 cycles at 50ºC) with the inner primers alone, only M. leprae DNA showed amplification. A combination of PS3/PS4 and NO3/NO4 in OTN PCR resulted in the amplification of DNA from M. leprae but not from the other mycobacterial species (Fig. 2b). Wc, therefore, concluded that this combination of primers was specific for M. leprae DNA under the conditions used. It should be noted that four closely migrating bands were seen with M. leprae, representing the products from the four possible combinations of primers.
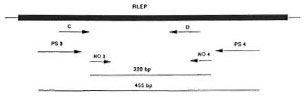
Fig. 1. Diagrammatic representation of the posi-tions of different RLEP primer pairs. PS3/PS4 and NO3/NO4 are outer and inner primers used in thisstudy and generate 455-bp and 320-bp fragments, respectively. Primers C and D were described elsewhere (20).
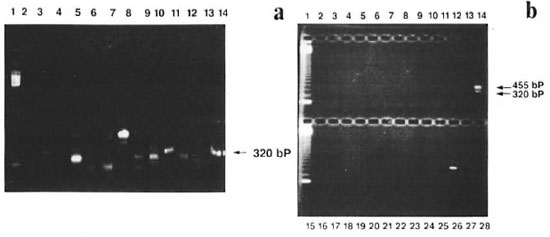
Fig. 2. Specificity of the primers; amplified products were analyzed on 1.8% agarose gel. a = Primers NO3/NO4 were used to amplify 1 ng of DNA from M. asiaticum (lane 2), M. avium (lane 3), M. xenopi (lane 4), M.fortuitum-peregrinum (lane 5), M. nonchromogenicum (lane 6), M. tuberculosis (lane 7), M. bovis (lane 8), M.tuberculosis (lane 9), M. ulcerans (lane 10), M. scrolidaceum (lane 11), M. flavescens (lane 12), M. smegmatis (lane 13), M. leprae (lane 14). Line 1 shows the 123-bp DNA ladder used as a size maker. b = A combinationof PS3/PS4 and NO3/NO4 were used to amplify 1 ng of DNA from lanes 2-14 (same as 2a), M. phlei (lane16), M. fortuitum-ranae (lane 17), M. gordonae (lane 18), M. fortuitum (lane 19), M. chelonei (lane 20), M.duvalli (lane 21), M. terrae (lane 22), M. vaccae (lane 23), M. marinum (lane 24), M. tuberculosis (lane 25), M. leprae (lane 26), negative controls (lanes 27 and 28). Lanes 1 and 15 show 123-6p DNA ladder used as size marker
The sensitivity of the PCR was determined by the amplification of genomic M. leprae DNA dilutions ranging from 1 ng to 100 ag. One fg of DNA was routinely detected (1 genome corresponds to 5 fg), a sensitivity considerably greater than we had found using a single-copy sequence, where sensitivity was between lOfgand 100 fg (9).
Colorimetrie detection of PCR products. Agarose gel electrophoresis is not a convenient assay for screening large numbers of samples. Wc therefore adapted the OTN PCR for colorimetrie detection. The internal primers NO3 and NO4 were labeled with digoxigenin (Dig) and biotin, respectively, at their 5' ends. OTN PCR therefore produced a 320-bp band which was labeled with a different ligand at each end. These molecules were captured in avidin-coated microtiter plate wells, and detected by incubation with alkaline phosphatasc-conjugatcd anti-Dig antibody, followed by color development in the presence of para -nitrophenol.
The sensitivity and specificity of the color assay was assessed using the same M. leprae and other mycobacterial DNA samples as before. There was complete concordance between the presence of bands on agarose gels and color production (not shown). An important feature of the colorimctric assay is that the background levels arc extremely low. The background readings observed with negative controls were in the range of 0 to 0.009. Hence the cutoff between the positive and negative samples was taken as the mean of negative controls (0.001) + 10 standard deviations (0.02). In practice, there was a clear distinction between positive and negative readings.
In order to determine the intra-assay variability in the color development, each PCR product was added to the wells in duplicate. Very little variation was observed between the optical readings of the same sample.
Detection of M. leprae DNA in frozen skin biopsies. The OTN PCR was next used to test clinical samples: 5- µ m sections from frozen biopsies taken from 40 untreated leprosy patients were processed and checked for the presence of inhibitors, and aliquots were then analyzed by OTN PCR. The diagnoses of the patients ranged across the whole leprosy spectrum. Four sections from each biopsy were tested, and amplified products were detected by agarose gel electrophoresis and color detection. Samples were counted as positive if M. leprae DNA was detected in any of the four sections. M. leprae DNA was detected in all the multibacillary sections (BI = 2 or above; N = 17). In paucibacillary patients, 69% of the samples (N = 13) with a BI of 1 to 1.5 and 70% of the samples (N = 10) with a BI of 0 were also positive (Fig. 3).
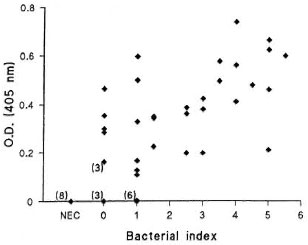
Fig. 3. Sensitivity of OTN PCR colorimetric method in detecting M. leprae from the skin biopsy sections of leprosy patients. Optical densities (ODs) of the samples from leprosy patients with varying bacterial in-dexes (0-5.5; N = 40) and nonendemie controls (NEC;N = 8) measured at 405 nm.
Not all sections from the same biopsy were positive. In the samples with the highest Bis (4-5.5), all sections were positive by PCR; in samples with Bis of 2-3.5, between two and four sections out of four were positive; and in samples with Bis of 0-1.5 only, one of the four sections was positive in most cases that showed amplification. All negative controls (N = 105, containing sterile distilled water instead of clinical sample) and eight skin-biopsy sections from patients with conditions other than leprosy remained negative.
Effect of chemotherapy. We have previously used limiting dilution PCR (LD-PCR) to quantitatc M. leprae DNA in sections of frozen skin biopsies. This study was used to determine the effect of MDT on M. leprae viability by following multibacillary patients for 2 years (9), hypothesizing that M. leprae DNA is broken down rapidly after the death of the organism. We found that in approximately half of the cases, M. leprae DNA became undetectable by PCR of the single copy pra gene (9). Since the RLEP PCR described here is more sensitive, we re-analyzed these samples (The Table). Using RLEP as a target, amplification was observed with some previously negative samples, although 7 out of 8 still became negative by 24 months. The one exception which stayed positive throughout had previously appeared anomalous, in that it was negative at 3, 6 and 12 months, but positive at 24; using RLEP, we detected M. leprae DNA at all time points. These results support our earlier findings, that the bacilli had all been killed by 24 months in half of the multibacillary patients, and that DNA is degraded so that even a multicopy repeated sequence becomes undetectable, while in other patients a small number of bacilli persist.
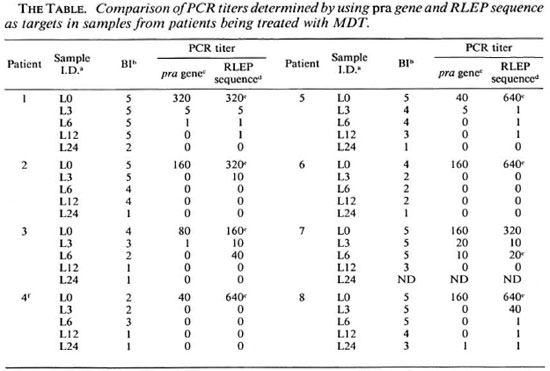
a Frozen skin sections collected from patients being treated with MDT at Marie Adelaide Leprosy Centre,Karachi.
b BI (bacterial index) = number of AFBs measured on a logarithmic scale.
c The greatest dilution at which amplification was observed using pra gene as a target; a titer of 1 indicatesthat amplification was observed only when the sample was run undiluted.
d The greatest dilution at which amplification was observed using RLEP sequence as a target; a titer of 1indicates that amplification was observed only when the sample was run undiluted.
e Highest dilution at which PCR was still positive.
f Patient was histologically characterized as indeterminate.
DISCUSSION
Although established multibacillary leprosy can be diagnosed easily using clinical examination and microscopy, the diagnosis of early disease, indeterminate forms, and cases in whom the bacilli are difficult to demonstrate, continues to be a problem. There is, therefore, a need to develop a sensitive and specific method for rapid and definitive identification of M. leprae in cases whose clinical and histological findings are equivocal. We have developed an OTN PCR for a sequence within the repetitive element of M. leprae . This sequence has been reported to be specific for M. leprae by hybridization (2). However, when using PCR, we found both nonspecific amplification and what appeared to be specific amplification, giving rise to bands of the expected sizes with some species. Since PCR can be effective with relatively small regions of homology compared to filter hybridization, these results are not incompatible. In order to verify the specificity, the two primer pairs were tested alone as well as in a combination. Some crossreactivity was observed with single sets of primers, but the nested reaction was specific. Clearly, if related sequences do exist in other species, their characterization and sequencing will allow more specific primers to be designed.
Since the repetitive element occurs in 28 copies in the genome, its use as a target has given the system an advantage over methods using single-copy targets in terms of sensitivity. Woods and Cole (20) have shown the RLEP sequence to be 15-fold more sensitive after PCR than a single copy sequence. The sensitivity of our assay has been consistently 1 fg, as determined by a colorimetric method as well as agarose gel analyses of purified M. leprae DNA. Colorimetric methods for the detection of M. leprae have been described by another group (15,16) with a detection limit of 100 fg to 125 fg of genomic DNA.
In previous studies using PCR based on pra (5) and RLEP (22) 56% and 73%, respectively, of samples with no detectable bacilli on microscopy were shown to be PCR-positive. Our results agreed with the previous reports, finding 70% of such samples to be positive, as well as 69% of samples with Bis of 1 -1.5. How can the 30% of PCRnegative samples be explained? A variety of factors can influence the PCR result of sample with Bis of 1 or 0; the area chosen for biopsy may not be representative of the lesion or bacilli, if present, had completely degraded DNA which could not be amplified. Patients may not have had leprosy or AFB seen on microscopy may not have been M. leprae. AFB related to M. scrofulaceum and M. avium have been isolated from the lesions of leprosy patients (6), although there is no significant evidence for false-positive smear results in leprosy caused by cultivable mycobacteria. Alternatively, the DNA extraction and amplification methodologies may need to be improved further.
In most paucibacillary samples, not all of the sections gave positive PCR results. This would not be surprising if the number of bacilli is very low. Other groups have pooled multiple sections prior to DNA extraction (22), or used a single section, but amplified a larger proportion of the extracted DNA (5).
In order to determine the efficacy of MDT in leprosy, quantitative OTN PCR was applied to the samples collected from patients prior to and at regular intervals after the start of MDT. The titers obtained were compared to those we had observed with the pra gene as a target. Our results showed that previously negative samples were PCRpositive, and the titers were generally higher with primers targeting the RLEP sequence. This could be due to the higher detection limit of the assay which was consistently 1 fg compared to 10 fg to 100 fg observed with the pra gene as a target. Any sample with intact M. leprae DNA <100 fg but >1 fg would therefore be detectable by an assay using RLEP sequence and not pra gene as a target. The other possibility could be that if some portion of DNA from the dead or decomposed M. leprae can serve as a suitable template, the likelihood of amplification would be higher with primers targeting a multicopy sequence. Although a relationship between positive PCR results and potentially viable M. leprae has been suggested by several workers (5,9,20), well controlled studies are needed to determine the exact relationship between viability and positive PCR signals. Another important issue that requires investigation is the time interval that elapses between the biological death of M. leprae and cither autolytic or host-induced degradativc changes in the bacillary DNA which result in the loss of signals for PCR. Studies by Williams, el al. (18) have shown that whereas the mouse foot pad assay indicated that M. leprae lose viability after only 2 weeks of treatment, the changes that rendered the M. leprae DNA nonamplifiablc required at least 2 months.
We and other investigators (9,18) have demonstrated previously that PCR could be used for monitoring the efficacy of drug therapy in leprosy. In the present study wc have again shown that potential by using a more-sensitive method. Relapse in the first documented case following MDT was caused by drug-sensitive pcrsistcrs rather than the development of drug resistance (4). The persisters, because of their dormant state, are protected from the effect of drugs and resume multiplication as soon as the drugs arc withdrawn. When MDT was introduced it was hypothesized that any post-MDT relapses would be due to the multiplication of drug-susceptible pcrsistcrs. Currently, the only reliable method available for the detection of persistcrs is growth in thymectomized and irradiated mice (3), which is not only expensive but is extremely time consuming. The use of PCR would be a cheap and rapid alternative.
The application of this PCR protocol to a limited number of clinical samples shows the potential of this assay for large-scale screening. The OTN PCR described here is relatively inexpensive, costing half as much as standard nested PCR, and uses detection equipment required for enzyme-linked immunosorbent assays (ELISAs) which are already available in many laboratories throughout the world, including those in developing countries.
Acknowledgment. We would like to thank Dr. Thomas Chiang, Dr. Qadccr Ehsan and Mr. Mustafa Khan for collection of samples; Dr. Peter Godfrey-Faussett for providing DNA samples from different mycobacterial species, and Dr. John Rayncs for help with the statistical analysis of the data. Sincere thanks are also expressed to Dr. Sebastian Lucas for histological classification of patients and to Dr. Hazel Dockrcll for coordinating the study. This investigation received financial support from LEPRA, ODA and the UNDP/ World Bank/WHO Special Programme for Research and Training (TDR).
REFERENCES
1. CHOU, Q., RUSSELL, M., BIRCH, D. E., RAYMOND, J. and BLOCH, W. Prevention of pre-PCR mispriming and primer dimcrization improves lowcopy-number amplifications. Nucleic Acids Res. 20(1992)1717-1723.
2. CLARK-CURTISS, J. E. and DOCHERTY, M. A. A species-specific repetitive sequence in Mycobacterium leprae DNA. J. Infect. Dis. 159(1989)7-15.
3. COLSTON , M . J. Application of the thymcctomized-irradiated mouse to the detection of persisting Mycobacterium leprae. Int. J. Lepr. 55(1987) 859-863.
4. CONSTANT-DESPORTES, M., GUELPA-LAURAS, C, CAROLINA, J., LEOTURE, A., GROSSET, J.-H. and SANSARRICQ, H. A case of relapse with drug-susceptible M. leprae after multidrug therapy. Int. J. Lepr. 59(1991)242-247.
5. DE WIT, M. Y. L., FABER, W. R., KRIEG, S. R., DOUGLAS, J. T., LUCAS, S. B., MONTREEWASUWAT, N., PATTYN, S. R., HUSSAIN, R., PONNIGHAUS, J. M., HARTSKEERL, R. A. and KLATSER, P. R. Application of a polymerase chain reaction for the detection of Mycobacterium leprae in skin tissues. J. Clin. Microbiol. 29(1991)906-910.
6. FANDINHO, F. C. O., SALEM, J. I., GONTUO-FILHO, P. F., MAROJA, M . F. and DAVID, H. L. Mycobacterial flora of the skin in leprosy. Int. J. Lepr. 59(1991)569-575.
7. HACKEL, C, HOWARD, S., PORTAELS, F., VAN EL-SEN, A., HERZOG, A. and BOLLEN, A. Specific identification of Mycobacterium leprae by the polymerase chain reaction. Mol. Cell Probes 4(1990)205-210.
8. HARTSKEERL, R. A., DE WIT, M. Y. L. and KLATSER , P. R. Polymerase chain reaction for the detection of M. leprae. J. Gen. Microbiol. 135(1989)2357-2364.
9. JAMIL, S., KEER, J. T., LUCAS, S. B., DOCKRELL, H.CHIANG, T. J., HUSSAIN, R. and STOKER, N. Use of polymerase chain reaction to assess efficacy of leprosy chemotherapy. Lancet 342(1993)264-268.
10. KEMP, D. J., CHURCHILL, M. J., SMITH, D. B., BIGGS, B. A., FOOTE, S. J., PETERSON, M. G., SAMARAS, N., DEACON, N. J. and DOHERTY, R. Simplified colori metric analysis of polymerase chain reactions: detection of HIV sequences in AIDS patients. Gene 94(1990)223-228.
11. PATTYN, S. R., URSI, D., LEVEN, M., RAES, V. and JAMET , P. Polymerase chain reaction amplifying DNA coding for species-specific rRNA of Mycobacterium leprae. Int. J. Lepr. 60(1992)234-243.
12. PLIKAYTIS, B. B., GELBER, R. H. and SHINNICK, T. M. Rapid and sensitive detection of Mycobacterium leprae using the nested-primer gene amplification assay. J. Clin. Microbiol. 28(1990) 1913-1917.
13. RIDLEY, D. S. and JOPLING, W . H. Classification of leprosy according to immunity; a five-group system. Int. J. Lepr. 34(1966)255-273.
14. SHEPARD, C. C. and MCRAE, D. H. A method for counting acid-fast bacteria. Int. J. Lepr. 36(1968)78-82.
15. VAN DER VLIET, G. M. E., DE WIT, M. Y. L. and KLATSER , P. R. A simple colorimetric assay for detection of amplified Mycobacterium leprae DNA. Mol. Cell Probes 7(1993)61-66.
16. VAN DER VLIET, G. M. E., HERMANS, C. J. and KLATSER , P. R. Simple colorimetric microtitcr plate hybridization assay for detection of amplified Mycobacterium leprae DNA. J. Clin. Microbiol. 31(1993)665-670.
17. WILLIAMS, D. L., GILLIS, T. P., BOOTH, R. J., LOOKER, D. and WATSON, J. D. The use of a specific DNA probe and polymerase chain reaction for the detection of Mycobacterium leprae. J. Infect. Dis. 162(1990)193-200.
18. WILLIAMS, D. L., GILLIS, T. P., FIALLO, P., JOB, C. K., GELBER, R. H., HILL, C. and IZUMI, S. Detection of Mycobacterium leprae and the potential for monitoring antileprosy drug therapy directly from skin biopsies by PCR. Mol. Cell Probes 6(1992)401-410.
19. WILSON, S. M., MCNERNEY, R., NYE, P. M., GODFREY-FAUSSETT, P. D., STOKER, N. G. and VOLLER , A. Progress toward a simplified polymerase chain reaction and its application to diagnosis of tuberculosis. J. Clin. Microbiol. 31(1993)776-782.
20. WOODS, S. A. and COLE, S. T. A rapid method for the detection of potentially viable Mycobacterium leprae in human biopsies: a novel application of PCR. FEMS Microbiol. Lett. 65(1989)305-310.
21. WOODS, S. A. and COLE, S. T. A family of dispersed repeats in Mycobacterium leprae. Mol. Microbiol. 4(1990)1745-1751.
22. YOON, K., CHO, S., LEE, S. M., ABALOS, R. M., CELLONA, R. V., FAJARDO, T. T., GUIDO, L. S., DELA CRUZ, E. S., WALSH, G. P. and KIM, J. Evaluation of polymerase chain reaction amplification of Mycobacterium leprae-specxixe repetitive sequence in biopsy specimens from leprosy patients. J. Clin. Microbiol. 31(1993)895-899.
1. M.Sc; Department of Clinical Sciences, London School of Hygiene and Tropical Medicine, Keppel Street, London WC1E 7HT, U.K.
2. Ph.D.; Department of Clinical Sciences, London School of Hygiene and Tropical Medicine, Keppel Street, London WC1E 7HT, U.K.
3. Ph.D., Department of Clinical Sciences, London School of Hygiene and Tropical Medicine, Keppel Street, London WC1E 7HT, U.K.
4. B.Sc, Department of Histopathology, University College, London Medical School, London, U.K.
5. Ph.D., Department of Microbiology, Aga Khan University, P.O. Box 3500, Karachi 74800, Pakistan.
Reprint requests to Dr. Stoker.
Received for publication on 17 May 1994.
Accepted for publication on 13 July 1994.