- Volume 62 , Number 1
- Page: 108–21
Immunology of leprosy: lessons f rom and for leprosy*
Editorial opillions expressed are those of the writers.
XIII LEPROSY CONGRESS STATE-OF-THE-ART LECTURE
We are pleased to have the opportunity of publishing the full texts of the Stateoft he-Art Lectures presented at the XIV International Leprosy Congress in Orlando, Florida, U.S.A., 29 August-4 September, 1993. The first two of these appear in this issue. Remaining lectures will appear in subsequent issues-RCH
It is my privilege to take you on a journey through the immunology of leprosy and itis an honor to be your travel guide.
The T-cell-mediated immune response involved in causing tuberculoid skin and plays a key role in leprosy and this is evident nerve pathology. from the leprosy spectrum. T-cell-dependent immunity to Mycobacterium leprae is high in healthy exposed individuals and in tuberculoid leprosy patients with localized disease, but is strikingly absent in lepromatous leprosy patients who have high bacillary loads and widely disseminated lesions. Thus, T-cell dependent immunity protects against dissemination of bacteria and of disease (Fig. 1). But for reasons thatare not entirely understood, cell-mediated immunity does not fully protect those individuals who develop tuberculoid leprosy. In fact the immune response may be directly involved in causing tuberculoid skin and nerve pathology.
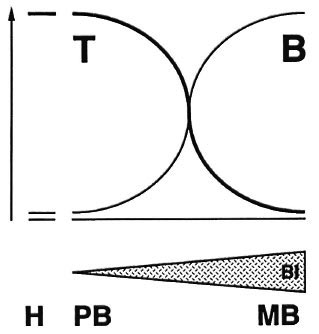
Fig. 1. The leprosy spectrum.
So the cellular immune response is a two-edged sword: it protects, by limiting bacillary growth, yet it also can harm: if turned on improperly, it can induce severe pathology like in tuberculoid leprosy and leprosy reactions. And if it is turned off improperly, bacteria can reach massive numbers and cause diffuse lepromatous pathology.
The question seems simple: How do we distinguish protective immunity from pathological immunity, and how do we induce one and avoid the other? Simple questions unfortunately almost never come with simple answers and leprosy is no exception,together with other infectious diseases such as tuberculosis, malaria, HIV and other diseases such as cancer and autoimmunity.
An incredible number of new discoveries in immunology have been made over the past few years, and they have crucial implications for our understanding of cellular immunity. In the classical view, cellular immunity was seen as a process in which macrophages take up bacteria and present them to T cells which then start to produce interferon-gamma (IFN- γ ) which helps the macrophage eliminate the intracellular parasite (Fig. 2).
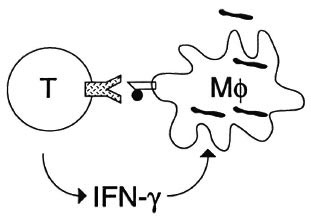
Fig. 2. Cell-mediated immunity; a classical view.
But we now know that the cellular immune response is much more complex (Fig. 3). We have learned about the many different antigens that mycobacteria express, about how bacteria invade into and are handled by the macrophage, how bacteria are able to escape or neutralize hostile compartments inside the macrophage. We are learning about how bacteria may switch on virulence genes uniquely within the infected host. We know how bacterial proteins are broken down into small peptide fragments which are then presented to T cells by specialized presenting molecules known as HLA. We know there are different peptide presentation routes and that these determine the type of T cell that is turned on. We know that there is not just a single helper-T cell involved in antibacterial immunity: there are many different types of T cells that diner in molecular markers, such as CD4 and CD8 and, more importantly, in immune functions such as their ability to kill various infected host cells and their ability to release different cytokines once they see antigen.
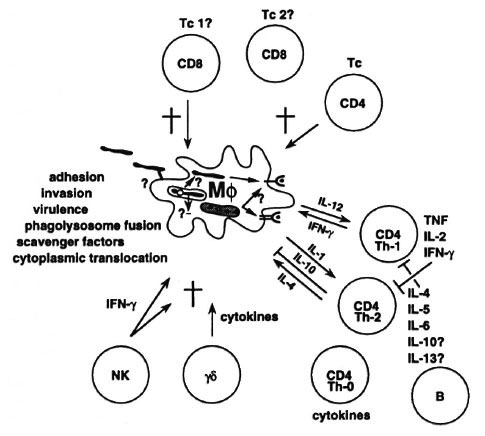
Fig. 3. Cell-mediated immunity; a 1993 view.
How does this relate to our understanding of leprosy, and eventually to the control and management of leprosy? I want to look at the various stages of the cell-mediated immune response in more detail and reflect about what we have learned, what we are learning, and what still needs to be learned.
M. leprae antigenic repertoire in cell mediated immune responses
Let us begin where leprosy begins, with M. leprae . A key question has always been which components of the bacterium are relevant to the immune system and which antigens induce protective immunity but avoid tuberculoid pathology. Is there a particular compartment within the leprosy bacillus where the important antigens come from, for example, are secreted antigens especially important or are cell-wall-bound antigens or cytoplasmic proteins or all of them? Many investigators have worked to address this question. As we heard in the lecture from Patrick Brennan, he together with Barry Bloom, Vijay Mehra and Robert Modlin has done important work from which we have learned that many human T cells react to proteins that copurify with the mycobacterial cell wall. Through this approach they have found that one protein particularly, called the M. leprae heat-shock protein 10 (hsp10), is often recognized by T cells, which could make it an interesting candidate for vaccines and skin tests.1 This is currently being further analyzed in human skin tests in Venezuela.
From our own studies, we find that a rather broad range of antigens appears to be seen by T cells.2, 3 Some T cells see heat-shock proteins such as hsp70, 65, 18 and 10; others react with secreted proteins like members of the fibronectin-binding protein family such as the 30-31 kDa proteins4 and the recently (in Jelle Thole's lab) defined M. leprae -specific 45 kDa protein;5 still others recognize as yet ill-defined antigenic fractions.2,3 But basically the total spectrum of antigens recognized by T cells is very wide. There is as yet no evidence for antigens that are exclusively recognized by patients or by healthy individuals so far.
Secreted proteins of mycobacteria
Much interest has focussed recently on proteins that are secreted by mycobacteria and that appear to be important targets for T cells. In most animal models of mycobacterial infection, protection against virulent mycobacterial species can be induced only by vaccination with live but not dead attenuated mycobacteria, even though both induce skin-test positivity. Because only live and not dead mycobacteria are able to secrete proteins, a possibility is that secreted proteins may be the major target proteins for protective immunity. Indeed, secreted antigens are seen extremely often by human T cells as reported by Huygen, Abou-Zeid, Harboe, Thole and many others (e.g.,4, 5).
Secreted antigens may be rapidly produced during the initial stages of infection and by their mere abundance elicit a rapid, early T cell response. But maybe they have other unique properties like, for example, the efficient entry of antigen-processing pathways, that make them highly immunogenic. The 30-31 kDa protein complex is particularly immunogenic, and recently three members of this protein family have been cloned for M. leprae and several were shown to stimulate T cells.
Although secreted antigens may appear to be major and perhaps even protective antigens, one has to keep in mind that live bacteria differ in many other aspects from dead bacteria other than in just the secretion of antigens. For example, live bacteria may enter other intracellular pathways in the macrophage, provide stronger adjuvant effects, switch on novel genes in the host, and so on. Each of these possibilities may be related to the ability of live bacteria to induce protective immunity. Be that as it may, it is evident that secreted antigens are a very important class of antigens.
An interesting study was recently published by Marchal and coworkers in Paris.6 They immunized mice with either live or dead BCG and then performed quantitative skin tests with limiting antigen dilutions. They found a novel, secreted protein that was recognized predominantly by live-BCG vaccinated animals. Its N terminal amino-acid sequence seems to be unique so far. Such approaches may provide important new information by focussing on a perhaps more relevant set of antigens, and may lead to the definition of better protecting antigens and the detection of virulence factors.
Differences in antigen recognition: role of HLA molecules
In our own studies, as I mentioned, we so far have found no antigens that are exclusively recognized by either leprosy patients or healthy exposed individuals. But we do see differences between different individuals in their ability to respond to particular proteins (e.g., 2). Mycobacterial antigens need to be presented at the macrophage surface before they can be recognized by T cells (Fig. 4). To that purpose macrophages have specialized presenting molecules called HLA molecules. HLA molecules bind small mycobacterial peptides that are produced in the endosomal-lysosomal compartment of the macrophage. HLA molecules then present these peptide fragments to T cells. Because there are many different HLA molecules, different individuals will differ in their HLA types, and because different HLA molecules bind different peptides, different individuals will react to different peptides and different proteins. For example, individuals who have a particular HLA type, DR3, appear to respond to a subset of M. leprae antigens, like hsp70, 65, 18 and the secreted 30-31 kDa fibronectin-binding protein family. There may be peptides in these proteins that DR3 prefers to bind with and conversely, DR3 may not bind peptides from other proteins such that no T-cell response can be generated.
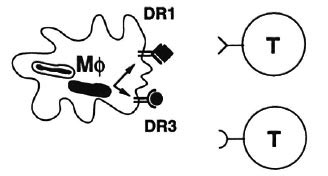
Fig. 4. Different HLA-DR molecules present different peptides to T cells.
We have indeed found peptides that only bind to DR3 and not to any other DR types. One example is the peptide coded by amino acids 3-13 of the hsp65 molecule which, as mentioned, is an important protein antigen for T cells.5 When we compared this peptide with other M. leprae peptides that are recognized by DR3+ individuals, all peptides appeared to look alike in that they have a common motif which is responsible for docking it into the DR3 molecule. Amino acids at particular positions are highly conserved and anchor the peptide into the polymorphic-peptide-binding site of the HLA-DR3 molecule.7 This may have implications for the design of (preventive or immunotherapeutical) recombinant vaccines, since an essential requirement is that each individual can respond to at least one of the proteins in the vaccine. Using such motifs, it may become possible to select particular proteins as good mycobacterial vaccine candidates which may be relevant now that the M. leprae genome is being sequenced entirely.
Genetic factors in leprosy
Genetic host-susceptibility factors. A question that I want to briefly discuss in this context is whether certain individuals are more susceptible to leprosy than others. There is strong and longstanding evidence that this is indeed the case, and it is not without reason that leprosy in the last centuries was believed to be inherited before Armauer Hansen discovered M. leprae . Nevertheless, only a few genetic factors have been identified so far.
One of these is encoded by HLA genes. Rene de Vries and colleagues were the first to find that HLA genes are important genetic factors in leprosy.8 It has become evident now that these HLA genes do not determine susceptibility to leprosy per se but, rather, control the type of leprosy that develops upon infection of susceptible individuals.
For example, among individuals susceptible to leprosy, those with HLA-DR3 more often develop tuberculoid leprosy whereas those with another HLA type, DQ1, more often develop lepromatous leprosy. Since different HLA molecules present different peptides from different proteins to T cells, HLA differences will lead to differences in T-cell responses. DR3 may induce a strong T-cell response and predispose to tuberculoid leprosy. DQ1 instead may induce a state of nonresponsiveness, perhaps through induction of M. leprae-specific suppressor cells, and thus predispose to lepromatous leprosy. A similar molecular mechanism has recently been demonstrated for severe malaria.9
But if HLA does not determine susceptibility to leprosy per se , what then does? Although we do not know the answer to that question at this moment, there is in mice a gene, called bcg , that appears to confer innate resistance to mycobacteria.10 This gene operates at the level of the macrophage. Very recently, a candidate gene has been cloned by Emile Skamene's group. This gene which is called "Nramp" probably encodes a macrophage-specific membrane transporter molecule. Once we learn what the gene does, we will be able to understand which protective mechanisms are affected in these susceptible mice and how such defects can be corrected. There is evidence for a similar gene in humans as well.
Mycobacterial virulence factors. The other side of genetics, or in other words the counterpart of genetic host-susceptibility factors, of course are genetic bacterial virulence factors. Because mycobacteria are not easy to work with, we know much more about virulence factors in other bacteria than mycobacteria, and many questions are unanswered. How do mycobacteria adhere to and invade into macrophages? (Fig. 5) Which molecules and cellular receptors are involved in adhesion and invasion, such as integrins, mannose receptors, growth factor receptors, fibronectin, antibodies, etc.? Do mycobacteria, for example, express a homolog of the invasin gene which is an important virulence factor in Yersinia species and which binds to integrins? How does target cell receptor activation relate to intracellular trafficking of mycobacteria? And how does that in turn control the resulting type of immunity or the macrophage's efficiency in handling the bacillus? And do mycobacteria interfere with signal transduction pathways in the target cell, for example, by the release of protein tyrosine kinases or phosphatases that are virulence factors in other bacteria? How do mycobacteria inhibit phagolysosome fusion, or scavenge reactive oxygen or nitrogen intermediates, or resist other unknown growth inhibiting mechanisms? And to what extent do mycobacteria literally exploit the immune response like Schistosoma mansoni which uses tumor necrosis factor-alpha (TNF- α ) to promote its own growth, or Trypanosoma species that do the same with IFN- γ . How do mycobacteria inside macrophages evade antigen presentation to CD4 T cells, enabling them to persist without being recognized?
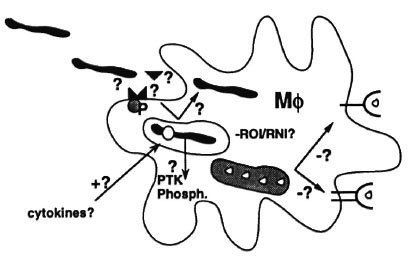
Fig. 5. Mycobacterial virulence factors.
It will be clear that we know almost nothing about the cell biology of intracellular infections and the "devious devices" that these parasites have adopted to survive or, even worse, to exploit the immune response to their own advantage.11, 12
For example, since we usually study bacteria in culture, we may have completely overlooked and consequently know nothing about an extremely important set of bacterial genes: namely, those bacterial genes that are uniquely and only switched on during infection of the host but not in culture. These genes may, of course, be major virulence genes. A major breakthrough in bacteriology has been the recent development of socalled in vivo expression technologies through which it is becoming possible to identify such genes.13 This technology unfortunately cannot yet be applied to mycobacteria, but many labs are working hard to achieve that goal. The identification of such genes may be of crucial importance for the development of new drugs, the understanding of how bacteria affect the host, and also why certain bacteria preferentially affect certain tissues, as M. leprae does with Schwann cells.
T cells in mycobacterial infections
Let's now leave antigens, macrophages, genetic and virulence factors, at least for a while, and stop at the next point of interest: the T cell, the director of the immune orchestra.
What do T cells do exactly? The classical model discussed before assumed that helper-T cells of the CD4-type recognize antigen plus HLA and then activate the macrophage through IFN- γ . However, as I pointed out as well, this simple model of cell-mediated immunity needs revision and the reality is much more complex than was anticipated.
Mycobacteria induce cytotoxic T cells
One lesson we have learned is that CD4 cells do not always "help" macrophages to eliminate mycobacteria. CD4 cells are also able to kill infected macrophages. Mycobacterium-pulsed macrophages but not control macrophages are readily killed by cytotoxic CD4 cells.14-17 CD4 cells are also able to kill other tissue cells from leprosy lesions, such as keratinocytes18 and melanocytes,19 once they have come in contact with mycobacteria.
Cytotoxic CD4 cells may mediate protection by several mechanisms (Fig. 6). One is by destroying the milieu in which M. leprae lives, namely, the macrophage, and M. leprae may simply be unable to survive outside of it. Also, when the cytotoxic cell kills the macrophage, it may at the same time also damage the M. leprae organisms inside that cell. Another mechanism is by killing burnt-out macrophages that are loaded with bacilli but have become refractory to activation. These bacilli can then be taken up again by fresh, activated macrophages that are much more potent killers of mycobacteria. Cytotoxic cells may thus reduce persistence and multiplication of bacilli. At the same time, it is easy to see that too extensive killing of tissue cells in leprosy lesions may lead to tissue damage, and the balance between protection and pathology may be rather delicate.
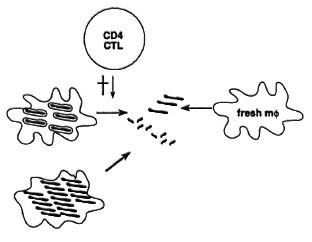
Fig. 6. Protective effects of cytotoxic T cells in mycobacterial infection.
T cells usually can be distinguished by the well-known CD4 and CD8 markers: they are either CD4 or CD8 positive. CD4 T cells sec peptides presented by HLA class II molecules like DR. CD8 T cells see similar peptides but they are presented by a different class of HLA molecules, namely, class I molecules. The basic difference is that class II molecules bind peptides from antigens in the endosomal/lysosomal pathway where bacteria are targeted; class I molecules instead bind peptides from the cytoplasm where, for example, viruses replicate. Hence, bacterium-specific T cells were thought to be mainly CD4, class-II restricted and virus-specific T cells mainly CD8, class-I restricted.
Tuna Mutis in our laboratory has recently found evidence however that mycobacteria not only induce CD4 T cells but also CD8 T cells, and that both T cells can kill macrophages that have taken up mycobacteria but they do not kill control macrophages. Such CD8 cells may be important with regard to protection, as shown in recent experiments with so-called knock-out mice. In these mice, a particular gene is specifically deleted or "knocked out" through homologous recombination. By this technique, mice were generated that do not express mouse-HLA class I molecules. As a consequence, these mice lack CD8 cells simply because CD8 cells were never expanded since there were no class-I molecules to drive their expansion. When these CD8-negativc mice were infected with Trypanosoma cruzi 20 or M. tuberculosis ,21 they rapidly died whereas similarly infected normal mice survived relatively long. This suggests that CD8 T cells are an important subset in protective immunity against virulent mycobacteria.
Paradoxically, we know virtually nothing about the role of CDS T cells in leprosy, about how mycobacteria enter the class-I antigen presentation pathway, how CD8 cells contribute to protective immunity, exactly when they appear in the immune response, and whether they are particularly important in pathology since many tissue cells express HLA class-I molecules and, thus, may be attacked by CD8 cells.
Many of these same questions also apply to so called gamma-delta ( γ-δ ) T cells. In addition to the CD4 and CD8 T cells, there is in fact a third T cell that expresses neither one of these markers. This third T cell also uses a slightly different form of receptor: whereas the common CD4 and CD8 T cells express an alpha-beta ( α β ) T-cell receptor, these T cells use the less common γ-δ variant. Without going into detail, I just want to say that several lines of evidence have suggested that γ-δ T cells might be important in antimycobacterial immunity. For example, γ-δ T cells rapidly accumulate and expand in lepromin skin tests22 as well as in mice early after infection with M. tuberculosis .
It is rather unclear what γ-δ cells see or do. Some evidence suggests that γ-δ T cells may recognize small molecular-weight components of mycobacteria; other evidence suggests that γ-δ cells may be involved in releasing cytokines, in regulating α β T cells, or in the killing of particular target cells. They may, in that respect, resemble nonspecific natural-killer cells that I have not talked about but which are also induced by mycobacteria and, as we have shown, efficiently kill infected macrophages in an antigen-independent, HLA-unrestricted fashion.14, 15
Cytokines and T cell subsets
We have looked at helper-T cells and cytotoxic cells, CD4 and CD8 T cells, α β and γ-δ cells, but one other critical issue is whatsoluble products T cells make. These products are called cytokines and are importantin mycobacterial infection.
In the past few years it has become clear that human CD4 T cells consist of functionally distinct subsets that differ in the cytokines they can produce. The subsets are called Th-1 for T helper 1 and Th-2 for T helper 2. Figure 7 briefly summarizes the main differences between the two subsets.23 Th-1 cells produce IL-2, a major T-cell growth factor; IFN γ , a major macrophage-activating factor, and TNF, which may be involved in granuloma formation. It follows that Th-1 cells are mainly involved in cell-mediated immune responses and, therefore, are relevant in mycobacterial infections. The opposite T-cell subset, Th-2, instead is mainly involved in regulating humoral immunity and releases another set of cytokines, notably IL-4 and IL-5, which regulate B cells, eosinophils and mast cells, and IL-6. Other factors such as IL-3 and GM-CSF are produced by both subsets. There are alsointermediate or mixed phenotypes of human CD4 T cells that are called Th-0 and that release both Th-1 and Th-2 cytokines.
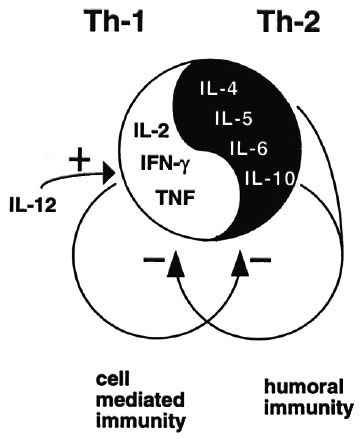
Fig. 7. T-helper 1 and T-helper 2 cells.
To add to the complexity, cytokines have not one but many functions, and also regulate other cells and cytokines. For example, Th-1 cytokines down-regulate Th-2 cellsand cytokines, and Th-2 cytokines down-regulate Th-1 cells and products. The effects of a particular cytokine therefore depend on the total local cytokine balance.
Cytokines and resistance to intracellularparasites
Mouse models. These discoveries have important implications for our understanding of cell-mediated immunity. In mice resistance to leishmaniasis depends on Th-1 cells whereas susceptible animals have a Th-2 response (reviewed in 23) (The Table). If the Th 1 cytokine IFN γ is neutralized with an antibody, a once-resistant animal now becomes susceptible to the same parasite and develops a Th-2 response. And the other way around, if you neutralize IL-4 in a susceptible animal, it now becomes resistant and develops a Th-1 response.

Cytokines and resistance to leprosy
The human situation. This, of course, has enormous implications in leprosy, tuberculosis and many other diseases. Could it be that leprosy is a disease with a disturbed cytokine balance in which at one pole Th-1 cells are activated that confer resistance whereas at the other pole, in lepromatous leprosy, Th-2 cells dominate that switch on humoral immunity but switch off cell-mediated immune responses? If that were the case, prevention and immunotherapy of leprosy would have to be directed at inducing or maintaining a correct cytokine balance.
Cytokines in tuberculoid leprosy. Let's look first at tuberculoid and then at lepromatous leprosy. Several laboratories have analyzed the cytokines that are produced by T cells from tuberculoid patients and healthy exposed individuals, in other words those well cell-mediated immunity to M. leprae . Some laboratories have looked directly at the cytokine products of M. leprae -rcactive T cells; others applied the powerful technique of the polymerase chain reaction (PCR) to detect cytokine-specific mRNA transcripts directly within leprosy lesions.
A consistent finding is that M. leprae -reactive T cells from tuberculoid patients are predominantly of the Th-1 phenotype: they produce high levels of IFN- γ and low or undetectable levels of IL-4.24-25 The induction of Th-1 cells is specific to mycobacteria because other antigens induce T cells with much lower IFN- γ /IL-4 ratios. The same pattern is seen when PCR products from tuberculoid lesions are analyzed on agarose gels. IL-2 and IFN- γ signals are particularly enriched in tuberculoid lesions.26
Gilla Kaplan injected Th-1-like cytokines such as IFN- γ and IL-2 locally in lepromatous leprosy lesions and saw clear signs of increased cell-mediated immunity accompanied by a significant increase in the degradation of M. leprae organisms.27 This again suggests that Th-1 cytokines are associated with bacterial elimination and, thus, protection in vivo .
Data from Ian Orme's group with mice where the gene for IFN- γ has been "knocked out" have confirmed that notion in an animal tuberculosis model.
So, cell-mediated immunity to mycobacteria is tightly associated with Th-1 cells and cytokines. If we want to induce cell-mediated immunity we will have to induce Th-1 cells. But even though it seems we have come closer to understanding cellular immunity, we certainly do not know all answers. For example, we see no difference in Th-1 cell activity between healthy exposed individuals and tuberculoid leprosy patients. So we cannot simply explain tuberculoid leprosy by a particular defect in cytokine production, at least at this point in time.
Cytokines in lepromatous leprosy. But what about lepromatous leprosy? Does the leprosy spectrum reflect the T-cell spectrum in that lepromatous T cells release Th-2 cytokines? Unfortunately, the results here are a little less clear.
First, Modlin and colleagues looked at in situ cytokine patterns in lesions by PCR and found that in lepromatous lesions mRNAs for typical Th-2 cytokines were enriched, notably IL-4, IL-5 and IL-10; whereas IFN- γ and IL-2 that were seen in tuberculoid lesions were absent in lepromatous lesions.26
Padmini Salgame studied cytokine production by T cells from lepromatous leprosy patients.28 Several T cells could specifically suppress the response of M. leprae -reactive helper-T cells. Thus, these M. leprae -specific suppressor-T cells could explain the characteristic T-cell unresponsiveness to M.leprae antigens in lepromatous patients. On balance, the results showed that suppressor-Tcells released more IL-4 than IFN γ ; where-as this was not seen for cells that did not suppress. Moreover, an antibody that neutralized the activity of IL-4 blocked suppression, suggesting that IL-4 was a crucial mediator in suppression (Fig. 8).
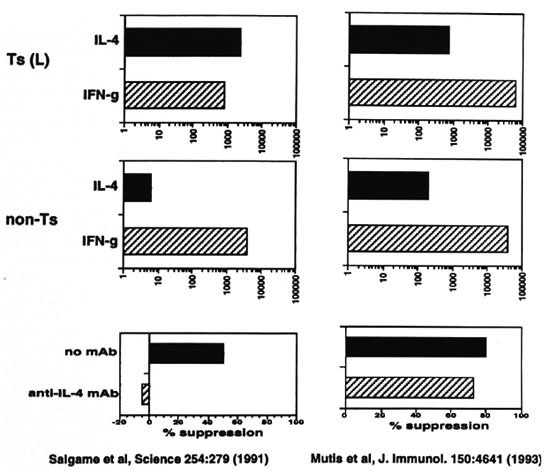
Fig. 8. Cytokine production by T cells from lepromatous leprosy patients.
We also have studied this issue with similar suppressor-T cells and nonsuppressive-T cells that were derived from the peripheral blood of three lepromatous leprosy patients25 (Fig. 8). Mutis, et al . found that these suppressor-T cells release significantly moreIFN γ than IL-4, although they do produce more IL-4 than tuberculoid T cells. There was no striking difference however between suppressive and nonsuppressive T cells. Moreover, anti-IL-4 antibody did not block suppression, suggesting that in this system of suppression IL-4 did not play a major role. Furthermore, in primary ex vivo cultures of blood lymphocytes neither anti-IL-4 or anti-IL-10 changed the state of T-cell nonresponsiveness to M. leprae ; whereas control T-cell responses to M. tuberculosis remained intact.
It is, of course, possible that there are differences between the two groups of lepromatous patients, the source from which the T cells came (lesions versus blood), the mechanisms of unresponsiveness and, maybe, other unknown variables that may account for the observed differences with regard to the role of I L-4 in nonresponsiveness. However, the question of whether cytokines are involved in nonresponsiveness in lepromatous leprosy remains important anddefinitely should be analyzed further. Weneed to know, for example, what cytokines prevent nonresponsiveness and turn on protective Th-1 cells, like perhaps IL-12. We also need to know whether cytokincs can and should be used as adjuvant immunotherapy in leprosy.
T-cell nonresponsiveness in lepromatous leprosy: further analysis
The defect in T-cell responsiveness to M. leprae in lepromatous leprosy is extremely specific. Responses to highly related bacteria like M. tuberculosis are intact even though they share many antigenic structures. What have we learned about the nature of this defect? How unresponsive are lepromatous leprosy patients really? It appears that it is rather easy to break through the nonresponsive state, at least in the test tube. When we challenge T cells from lepromatous patients with whole M. leprae , we observe the typical state of nonresponsiveness (Fig. 9). But if the same T cells are stimulated with purified, individual antigens or gel-separated antigen fractions, they appear to respond rather well toward those M. leprae antigens.3, 29, 30 (and unpublished data). This is a consistent observation in our hands that applies to quite a number of other antigens as well. The implication is that there is "something" in M. leprae that suppresses immunity and that if we take that something away without knowing what it is, immunity starts to reappear.
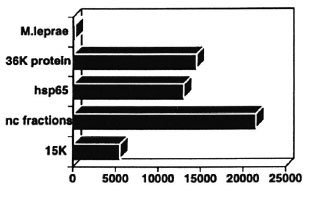
Fig. 9. T cells from unresponsive BL/LL patients respond to individual M. leprae antigens.
The preliminary vaccine trial results in Venezuela show that the combined vaccine of BCG with killed M. leprae does not protect much better against lepromatous leprosy than BCG alone.31 But if in susceptible individuals M. leprae somehow actively turns off the protective immune response, a combined vaccine of BCG and whole M. leprae may, indeed, not work better than BCG alone. We may need to identify M. leprae antigens that are able to trigger M. leprae -specific T cells in lepromatous patients without inducing nonresponsiveness. And such antigens may be already at hand, as we just saw. Such antigens would then be ideal candidates to incorporate, for example, in recombinant BCG vaccines both for prevention and perhaps adjuvant immunotherapy of lepromatous leprosy (Fig. 10).
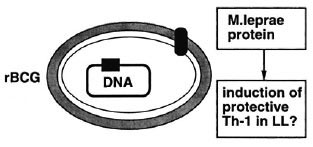
Fig. 10. Recombinant BCG: immunoprevention and immunotherapy of multibacillary leprosy.
Bill Jacobs, Barry Bloom, Rick Young, Ken Stover and others have pioneered the development of new techniques through which the stable, high expression of foreign genes can be achieved in BCG. Various antigens, like HIV proteins, have been expressed now in BCG and specific immunity can be achieved in mice with these new vaccines. The results are very promising sinceall arms of the immune system are activated: CD4 and CD8 T cells, Th- 1 -like helper cells, cytotoxic-T cells and also antibodyproduction is seen.32, 33 So, this new generation of vaccines may provide useful new tools for the prevention and, perhaps, immunotherapy of leprosy.
Through the work of Stover and colleagues, we are also beginning to understand where in BCG such antigens need to be expressed:34 when a major protein of Borrelia burgdorferi, the bacterium that causes Lyme disease, was expressed as a membrane-bound lipoprotein, it induced protective antibody responses. Interestingly, this was not the case when the very same antigen was expressed as a cytoplasmic or secreted protein in BCG.
Leprosy reactions
But perhaps the biggest problem of all in leprosy management and control is the occurrence of leprosy reactions. Cell-mediated immune responses have long been suspected to be involved in the pathogenesis of reversal reactions. Robert Modlin and colleagues have analyzed cytokine patterns in lesions of type 1 and type 2 reactions and basically found that Th-1-like cytokine signals tended to predominate in reversal reactions whereas Th-2-like cytokines were enriched in erythema nodosum leprosum (ENL).35 From Gilla Kaplan we know that TNF- α and IL-6 production are greatly increased in ENL patients.3 6 Thus, the immunopathology of type 1 and type 2 reactions is associated with quite different cytokine patterns.
We already have looked at the regulatory interactions in the cytokine network. Th-1 cytokines inhibit Th-2 cytokines, and vice versa. Because cytokines are such important regulators they may provide a new form of immunotherapy for leprosy reactions, cmploying inhibitory cytokines or antibodies, and this is a possibility that needs consideration.
But what triggers acute neuritis? Why are Schwann cells damaged? Do Schwann cells present M. leprae or self peptides, or are they killed as bystanders in inflammatory reactions? Or does M. leprae directly affect Schwann cell metabolism? Birhane Kale Ab has recently isolated T cells from acute neuritis lesions from routinely taken, diagnostic biopsies. The data are still preliminary but he is seeing two important things: first, many of the T cells from nerve lesions do indeed respond to M. leprae , thus for the first time providing a direct link between M. leprae responsiveness and neuritis. Secondly, several of the T cells also react to particular nerve-associated antigens. We do not know exactly what antigens are seen, whether it is the same T cell that recognizes both antigens or whether these are different T cells, what type of T cells and cytokines are involved but, whatever the answers, such T cells may help us to understand what is happening inside neuritis lesions and thus give us tools to specifically prevent and hopefully treat neuritis.
We have come to the end of our tour through the immunology of leprosy and have seen several objects of interest, albeit maybe from a distance. To be sure, we have learned much from leprosy for immunology. But have we also learned from immunology for leprosy? On the one hand, as long as we do not understand exactly what protection is, how different T-cell subsets contribute, how cytokine networks, virulence factors, macrophage bactericidal mechanisms, and so on, contribute, we cannot conclude with certainty that we have learned lessons from immunology for leprosy. But, at the same time, we are learning lessons for leprosy. And these lessons may lead to ways to induce protection, to vaccines that prevent lepromatous leprosy. Or when we discover more about virulence factors and genes that mycobacteria switch on during infection, these may be targeted with specific drugs. Or we may be able to design effective cytokine-based immunotherapy for reversal reactions or, when we define the target antigens, design tools for early detection and prevention of reactions. But we need to learn much more from and for leprosy. The questions we have to deal with are as difficult in leprosy as they are in other infectious diseases such as tuberculosis, malaria, HIV, autoimmune diseases and cancer.
Data from India suggest that 7 or 8 years of multidrug therapy (MDT) have had no significant impact on the incidence of leprosy.37 If this is true, the implication is clear: contrary to current beliefs MDT may not reduce the transmission of leprosy. We must not and may not make the same mistake twice and we had better learn from tuberculosis. Until we are sure we can manage leprosy we have to search for a better understanding of leprosy and its immunology so that we can put our lessons in practice for leprosy.
- Tom H. M. Ottcnhoff, M.D., Ph.D.
Department of Immunohematology and Bloodbank
University Hospital Leiden
2333 AA Leiden, The Netherlands
1. Mehra, V., Bloom, B. R., Bajardi, A. C.,'Grisso, C. L., Sidling, P. A., Alland, D., Convit, J., Fan, X., Hunter, S. W., Brennan, P. J., Rca T. H. and Modlin, R. L. A major T cell antigen of Mycobacterium leprae is a 10-kD heat shock cognate protein. J. Exp. Med.175(1992)275-284.
2. Janson, A. M., Klatser, P. R., van der Zee, R., Cornelisse, Y. E., de Vries, R. R. P., Thole, J. E. R. and Ottenhoff, T. H. M. A systematic molecular analysis of the T cell stimulating antigens from Mycobacterium leprae with T cell clones of leprosy patients. Identification of a novel M. leprae hsp 70 fragment by M. leprae -specific T cells. J. Immunol. 147(1991)3530-3537.
3. Ottenhoff, T. H. M., Converse, P. J., Gebre, N.,Wondimu, A., Ehrenberg, J. P. and Kiessling, R. T cell responses to fractionated Mycobacterium leprae antigens in leprosy. The lepromatous nonresponder defectcan be overcome in vitro by stimulation with fractionated M. leprae components. Eur. J. Immunol. 19(1988)707-713.
4. Thole, J. E. R., Schiiningh, R., Janson, A. A. M., Garbe, T., Cornelisse, Y. E., Clarck-Curtiss, J. E., Kolk, A. H. J., Ottenhoff, T. H. M., de Vries, R. R. P. and Abou-Zeid, C. Molecular and immunological analysisof a fibronectin binding, secreted antigen of Mycobacterium leprae . Molec. Microbiology 6(1992)153-163.
5. Wielis, B., van Agterveld, M., Janson, A. A. M., Clark-Curtiss, J., Rinke De Wit, T., Harboe, M. and Thole, J. E. R. Characterization of a Mycobacterium leprae antigen related to the secreted Mycobacterium tuberculosis protein MPT 32. (submitted for publication).
6. Romain, F., Angier, J., Pescher, P. and Marchal, G. Isolation of a proline-rich mycobacterial protein eliciting delayed-type hypersensitivity reactions only in guinea pigs immunized with living mycobacteria.Proc. Natl. Acad. Sci. U.S.A. 90(1993)5322-5326.
7. Geluk, A., van Meijgaarden, K. E., Janson, A. A. M., Drijfhout, J-W., Meloen, R., de Vries, R. R. P. and Ottenhoff, T. H. M. Functional analysis of DR17(DR3)-restricted mycobacterial T cell epitopes reveals DR17 binding motif and enables the design of allele-specific competitor peptides. J. Immunol. 149(1992)2864-2871.
8. de Vries, R. R. P. and Ottenhoff, T. H. M. Immunogenics of leprosy. In: Leprosy. 2nd edn. Hastings, R. C., ed. Edinburgh: Churchill Livingstone. (in press).
9. Hill, A. V. S., Elvin, J., Willis, A. C., Aidoo, M., Allsop, C. E. M., Gotch, F. M., Gao, X. M., Takiguchi, M., Greenwood, B. M., Townsend, A. R. M., McMichael, A. J. and Whittle, H. C. Molecular analysis of the association of HLA-B53 and resistance to severe malaria. Nature 360(1992)434-439.
10. Vidal, S. M., Malo, D., Vogan, K., Skamene, E. and Gros, P. Natural resistance to infection with intracellular parasites: isolation of a candidate for bcg .Cell 73(1993)469-485.
11. Sher, A. Parasitizing the immune system. Nature 356(1992)565-566.
12. Portnoy, D. A. and Smith, G. A. Devious devices of Salmonella . Nature 357(1992)536-537.
13. Mahan, M. J., Slauch, J. M. and Mekalanos, J. J. Selection of bacterial virulence genes that are specifically induced in host tissues. Science 259(1993)686-688.
14. Kaleab, B., Ottenhoff, T., Converse, P., Halapi, E., Tadesse, G., Rottenberg, M. and Kiessling, R. Mycobacterial induced cytotoxic T cells as well as non-specific killer cells derived from healthy individuals and leprosy patients. Eur. J. Immunol. 20(1990)2651-2659.
15. Kaleab, B., Kiessling, R., van Embden, J. D. A., Thole, J. E. R., Kumararatne, D. S., Pisa, P., Wondimu, A. and Ottenhoff, T. H. M. Induction of antigen specific CD4+ HLA-DR restricted cytotoxic T lymphocytes as well as nonspecific nonrestricted killer cells by the recombinant mycobacterial 65 kilodalton heat shock protein. Eur. J. Immunol. 20(1990)369-377.
16. Mutis, T., Cornelisse, Y. E. and Ottenhoff, T. H. M. Mycobacteria induce CD4+ T cells that are cytolytic and display Thl-like cytokine secretion profile. Heterogeneity in cytotoxic activity and cytokine secretion levels. Eur. J. Immunol. (in press).
17. Ottenhoff, T. H. M. and Mutis, T. Specific killing of cytotoxic T cells and antigen-presenting cells by CD4+ cytotoxic T cell clones. A novel potentially immunoregulatory T-T cell interaction in man. J. Exp. Med. 171(1990)2011-2024.
18. Mutis, T., de Bueger, M., Bakker, A. and Ottenhoff, T. H. M. HLA class II human keratinocytes present Mycobacterium leprae antigens to CD4+ cytotoxic and proliferative T cells. Scand. J. Immunol. 37(1993)43-51.
19. Le Poole, I. C., Mutis, T., van den Wijngaard, J.G. J., Westerhof, W., Ottenhoff, T. H. M., de Vries,R. R. P. and Das, P. K. A novel, antigen presentingfunction of melanocytes and its possible relation tohypopigmentary disorders. J. Immunol. (submitted for publication).
20. Tarleton, R. L., Koller, B. H., Latour, A. and Postan. M. Susceptibility of beta-2 microglobulin deficient mice to Trypanosoma cruzi infection. Nature 356(1992)338-340.
21. Flynn, J. L., Goldstein, M. M., Triebold, K. J., Koller, B. and Bloom, B. R. Major histocompatibility complex class I restricted T cells are required for resistance to Mycobacterium tuberculosis infection. Proc. Natl. Acad. Sci. U.S.A. 89(1992)12013-12017.
22. Modlin, R. L., Pirmez, C., Hoffman, F. M., Torigian, V., Uyemura, K., Rea, T. H., Bloom, B. R. and Brenner, M. B. Lymphocytes bearing antigen specific γ/δ receptors accumulate in human infectious disease lesions. Nature 339(1989)544-548.
23. Powrie, F. and Coffman, R. L. Cytokine regulation of T-cell function: potential for therapeutic interven-tion. Immunol. Today 14(1993)270-274.
24. Haanen, J. B. A. G., de Waal-Malefijt, R., Res, P.C. M., Kraakman, E. M., Ottenhoff, T. H. M., de Vries, R. R. P. and Spits, H. Selection of a human T helper type I-like T cell subset by mycobacteria. J. Exp. Med. 174(1991)583-592.
25. Mutis, T., Kraakman, E. M., Cornelisse, Y. E., Spits, H., de Vries, R. R. P. and Ottenhoff, T. H. M. Analysis of cytokine production by mycobacterium reactive T cells. Failure to explain Mycobacterium leprae specific nonresponsiveness of peripheral blood T cells from lepromatous leprosy patients. J. Immunol. 150(1993)4641-4651.
26. Yamamura, M., Uyemura, K., Deans, R. J., Weinberg, K., Rea, T. H., Bloom, B. R. and Modlin, R. L. Defining protective responses to pathogens: cytokine profiles in leprosy lesions. Science 254(1991)277-279.
27. Kaplan, G., Kiessling, R., Hancock, G., Teklemariam, Sabawork, Sheftel, G., Job, C. K., Converse, P., Ottenhoff, T. H. M., Becx-Bleumink, M., Dietz, M. and Cohn, Z. A. The reconstitution of cell mediated immunity in the cutaneous lesions of lepromatous leprosy by recombinant interleukin 2. J. Exp. Med. 169(1989)893-907.
28. Salgame, P., Abrams, J. S., Clayberger, C., Goldstein, H., Convit, J., Modlin, R. L. and Bloom, B. R. Differing lymphokine profiles of functional subsets of human CD4 and CD8 T cell clones. Science 254(1991)279-282.
29. Ottenhoff, T. H. M., Wondimu, A. and Reddy, N. N. B. A comparative study on the effects of rlL-4, rIL-2, rIFN γ and rTNF- α on specific T cell non responsiveness to mycobacterial antigens in lepromatous leprosy patients in vitro . Scand. J. Immunol. 31(1990)553-565.
30. Sela, S., Thole, J. E. R., Ottenhoff, T. H. M. and Clark-Curtiss, J. E. Identification of Mycobacterium leprae antigens from a cosmid library: characterization of a 15 kilodalton antigen that is recognized by both the humoral and the cellular immune systems in leprosy patients. Infect. Immun. 59(1991)4117-4124.
31. Convit, J., Sampson, C., Zuniga, M., Smith, P. G., Plata, J., Silva, J., Molina, J., Pinardi, M. E., Bloom, B. R. and Salgado, A. Immunoprophylactic trial with combined Mycobacterium leprae/ BCG vaccine against leprosy: preliminary results. Lancet 339(1992)446-450.
32. Stover, C. K., de la Cruz, V. F., Fuerst, T. R., Burlein, J. E., Benson, L. A., Bennett, L. T., Bansal, G. P., Young, J. F., Lee, M. H., Hatfull, G. F., Snapper, S. B., Barletta, R. G., Jacobs, W. R. and Bloom, B. R. New use of BCG for recombinant vaccines. Nature 351(1991)456-460.
33. Aldovini, A. and Young, R. A. Humoral and cell-mediated immune responses to live recombinant BCG-HIV vaccines. Nature 351(1991)479-482.
34. Stover, C. K., Bansal, G. P., Hanson, NI. S., Burlein, J. E., Palaszynski, S. R., Young, J. F., Koenig, S.,Young, D. B., Sadziene, A. and Barbour, A. G. Protective immunity elicited by recombinant Bacille Calmette-Guêrin (BCG) expressing outer surface protein A (OspA) lipoprotein: a candidate Lyme disease vaccine. J. Exp. Med. 178(1993)197-209.
35. Yamamura, M., Wang, X-H., Ohmen, J. D., Uyemura, K., Rea, T. H., Bloom, B. R. and Modlin, R. L. Cytokine patterns of immunologically mediated tissue damage. J. Immunol. 149(1992)1470-1475.
36. Moreira, A. L., Sampaio, E. P., Zmuidzinas, A., Frindt, P., Smith, K. A. and Kaplan, G. Thalidomide exerts its inhibitory action on tumot necrosis factor alpha by enhancing mRNA degradation. J. Exp. Med. 177(1993)1675-1680.
* This State-of-the-Art Lecture in Microbiology was presented on 30 August 1993 at the XIV International Leprosy Congress in Orlando, Florida, U.S.A.